Здравствуйте, подскажите пожалуйста что такое эктопия миндалин мозжечка?
Миндалины мозжечка - это нижняя область мозжечка. В норме миндалины мозжечка располагаются выше большого затылочного отверстия. Эктопия миндалин мозжечка - это наиболее распространенная разновидность аномалий мозжечка (мальформация Киари I взрослы тип). Эктопия миндалин мозжечка представляет собой комплекс симптомов, указывающих на одно- либо двусторонее опущение миндалин мозжечка через большое затылочное отверстие в позвоночный канал. Развитию эктопии миндалин мозжечка могут способствовать различные аномалии развития данного органа. Важнейшим обстоятельством при развитии данной мальформации является тот факт, что клинические проявления, то есть симптомы, возникают у пациентов только после тридцати и даже сорока лет. Не менее важно и то, что эктопия миндалин мозжечка, которая отличается бессимптомным характером, не требует лечения, являясь случайно обнаруженной в результате магнитно-резонансной томографии. Лечение проводится только при проявлении симптомов и носит только оперативный характер. О рациональности проведения хирургического вмешательства судить врач.
Полезные статьи
Женское сердце с точки зрения кардиологииУ каждого свой взгляд на женское сердце, у каждого свой ключик к нему. Но даже самое каменное женское сердце может болеть и при этом, совсем не от любви. И тогда самым правильным является взгляд на него врача-кардиолога – уж он точно знает о женском серде все!
Приоритетные направления диагностики и интенсивной терапии респираторного дистресс-синдрома у больных сепсисом.Многочисленные исследования отражают всю сложность трактовки концепции развития респираторного дистресс-синдрома (РДС), которая бурно продолжает обогащаться новыми данными. Именно легкие в критических ситуациях наиболее...
Дневная сонливость, нарушения сна и ночная дыхательная недостаточность в практике терапевта и кардиолога (2 часть)Наиболее важным в диагностике этих состояний является правильная интерпретация и выявление состояния "избыточной дневной сонливости" - патологического желания спать, с которым пациентам приходится бороться. К сожалению, эта "борьба" будет проиграна и в большинстве случаев сон застанет человека в самый неподходящий момент (например, за рулем автомобиля), что может привести к настоящей драме.
С гипертонией можно и нужно жить.«Познание болезни есть уже половина лечения» К гипертонии относится так называемое правило трех половин. Из общего числа больных ГБ знает об этом лишь половина. Из тех, кто знает, лечится только половина пациентов. И правильно лечится опять-таки половина. Есть один путь исправления этой странной закономерности - просвещение граждан.
Аритмии сердцаПроблема нарушений сердечного ритма остаются постоянно актуальной – и сколько бы ни было работ, посвященных этой теме, исчерпать ее невозможно. Нарушения сердечного ритма повседневно встречаются во врачебной практике, спектр сердечных аритмий в этиологическом, клиническом, диагностическом, прогностическом плане чрезвычайно широк.
Выраженность коронарного атеросклероза у пациентов с электрокардиографическими признаками миокардиальной ишемии при холтеровском мониторировании.Цель работы – оценка специфичности диагностики ишемии миокарда с помощью холтеровского мониторирования ЭКГ и попытка клинико-коронароангиографического сопоставления у пациентов с ишемической болезнью сердца.
Работа и кровяное давление.У японцев есть специальное слово: Karoshi, оно означает смерть от переработки. Для многих из нас работа – главный источник стресса, и общепринято, что те счастливчики, которые имеют работу, работают все тяжелее и тяжелее. Это приводит к тому, что становятся всё актуальнее вопросы связанные с влиянием работы на кровяное давление.
Инфекционный эндокардит. Особенности течения, критерии диагноза, дифференциальная диагностикаСовременный инфекционный эндокардит (ИЭ) характеризуется полиэтиологичностью, обусловленной широким спектром микроорганизмов. Мировой опыт позволяет выделить определенные особенности клинической картины заболевания...
Насколько непогрешима генетическая коррекция?Сообщество ученых-медиков разделилось по вопросу о том, следует ли одобрить повсеместное введение генетической коррекции – метода, позволяющего определить, заложена ли в оплодотворенную яйцеклетку предрасположенность к генетическим заболеваниям.
Что такое грудная жаба?Грудной жабой в России называют стенокардию – один из вариантов ишемической болезни сердца. При стенокардии сердце болит и ноет, потому что сердечной мышце не хватает кислорода из за нарушения кровообращения в коронарных артериях. Об этом заболевании рассказывают в этой статье специалисты в кардиологии Посольства медицины.
Что на самом деле вредно для нашего сердца?На самые распространённые вопросы о здоровье главного органа в организме ответили в своей книге известные британские кардиологи Роберт и Дебора Асхайм.
ГЕНЫ ЭНДОТЕЛИАЛЬНЫХ ФАКТОРОВ И АРТЕРИАЛЬНАЯ ГИПЕРТОНИЯ
Здоровое сердце в одной таблетке?Медики утверждают, что, прописав один-единственный препарат всем людям старше 55 лет, можно будет на 80% процентов сократить число сердечных заболеваний в развитых странах.
Диета, рекомендуемая европейским обществом по изучению атеросклероза Ишемическая болезнь сердца – из века в век…Мало, кто из нас, никогда не испытывал неприятного покалывания в отсиженной ноге или отлежанной руке. Связано это с тем, что, находясь в «прижатом» положении, конечность испытывает дефицит в притекающей к ней крови и, поступающем вместе с ней, кислороде, жизненно необходимых, каждому органу.
Гипертонический кризГИПЕРТОНИЧЕСКИЕ КРИЗЫ возникают при гипертонической болезни. В большинстве случаев характеризуются сочетанием системных и регионарных, преимущественно церебральных, ангиодистоний, тип и соотношение которых в каждом...
Артериальная гипертония у больных сахарным диабетом 2 типа и метаболическим синдромомПроблема артериальной гипертонии (АГ) в сочетании с метаболическим синдромом (МС) и сахарным диабетом 2 типа (СД) находится в центре внимания ученых всего мира в связи с повышенным риском развития сердечно-сосудистых осложнений...
Головная больСердце - качает кровь, кишечник - занимается пищеварением, у каждого своя функция. А вот голове приходится и смотреть, и слышать, и говорить, и при этом, еще и думать! Тут есть от чего заболеть.
Термин «мальформация Киари» является предпочтительным по сравнению с традиционным «мальформация Арнольда-Киари» в связи со значительно большим вкладом, внесенным Киари.
Мальформация Киари состоит из 4 типов аномалий заднего мозга, вероятно не связанных между собой. Большинство случаев приходится на типы 1 и 2 (см. табл. 6-5). На остальные варианты приходится крайне ограниченное количество случаев.
Табл. 6-5. Сравнительная характеристика 1-го и 2-го типов мальформации Киари (с изменениями)
Мальформация Киари 1-го типа
Т.н. первичная эктопия мозжечка. Это редкая аномалия, при которой имеется только каудальное смещение мозжечка с вклинением миндалин ниже БЗО (критерии см. МРТ ниже) и «удлинением миндалин типа затычки». В отличие от 2-го типа продолговатый мозг не смещен каудально (некоторые авторы с этим не согласны), ствол мозга не вовлечен, нижние черепные нервы не удлиненны, верхние шейные нервы не имеют направления в сторону головы.
Может быть фиброз мягкой и арахноидальной оболочек вокруг ствола мозга и миндалин. Могут быть гидромиелия и сирингомиелия СМ. Некоторые случаи могут быть приобретенными, после люмбо-перитонеального шунтирования или множественных (травматичных) ЛП.
Эпидемиология
Средний возраст клинического проявления 41 год (пределы: 12-73 года). Наблюдается некоторое преобладание & (&:%=1,3:1). Средняя продолжительность симптоматики определенно связанной с мальформацией Киари составляет 3,1 года (пределы: 1 мес – 20 лет). Если включить жалобы, которые носят неспецифический характер, напр., Г/Б, то этот срок составит 7,3 года. С наступлением эры МРТ эта задержка диагностики, возможно, станет меньше.
Клинические проявления
У пациентов с мальформацией Киари могут быть любые или даже все из следующих симптомов:
1. сдавление ствола мозга на уровне БЗО
2. ГЦФ
3. сирингомиелия
4. разделение внутричерепного отдела от спинального с периодическими повышениями ВЧД
Жалобы
Наиболее частой жалобой является боль (69%), особенно Г/Б, которая обычно ощущается в затылочной области (табл. 6-6).
Табл. 6-6. Жалобы при мальформации Киари 1-го типа (71 больной)

Г/Б часто вызывается разгибанием шеи и пробой Вальсальвы. Слабость также достаточно выражена, особенно при одностороннем пожимании руки. Может наблюдаться симптом Лермитта. Вовлечение нижних конечностей обычно проявляется двусторонней спастикой.
Симптомы
См. табл. 6-7. Три основных группы симптомов:
1. синдром сдавления в БЗО (22%): атаксия, кортико-спинальные и чувствительные нарушения, мозжечковые симптомы, паралич нижних ЧМН. 37% больных имеют тяжелые Г/Б
2. синдром центрального поражения СМ (65%): диссоциированные чувствительные нарушения (потеря болевой и температурной с сохраненной поверхностной и глубокой чувствительностью), иногда сегментарная слабость и признаки поражения длинных проводящих путей (сирингомиелический синдром). В 11% случаев наблюдается паралич нижних ЧМН
3. мозжечковый синдром (11%): атаксия туловища и конечностей, нистагм, дизартрия
Табл. 6-7. Симптомы при мальформации Киари 1-го типа (121 больной)

* в классическом случае: нистагм вниз при вертикальных движениях или ротаторный нистагм при горизонтальных движениях; также включает осциллопсию
Направленный вниз нистагм считается характерным для этого заболевания. В 10% случаев неврологический статус пациентов без изменений с единственной жалобой на затылочную Г/Б. В некоторых случаях основной жалобой может быть спастика.
Естественное течение
Естественное течение точно неизвестно (имеется только 2 публикации на эту тему). Пациент может оставаться в стабильном состоянии в течение нескольких лет с периодическими ухудшениями. В некоторых случаях возможно спонтанное улучшение (оспаривается).
Диагностика
Обзорные краниограммы
При анализе 70 обзорных краниограмм изменения были обнаружены только в 36% случаев (в 26% случаев была базиллярная импрессия, в 7% - платибазия; по одному пациенту имели болезнь Пэджета и вогнутый скат). Из 60 спондилограмм патологические изменения были в 35% случаев (включая ассимиляцию атланта, расширение канала, сращения шейных позвонков, агенезию задней дуги атланта).
Диагностический метод выбора. На МРТ легко обнаружить большинство из выше указанных аномалий, включая вклинение миндалин, а также гидросирингомиелию, которая наблюдается в 20-30% случаев. Также видна вентральная компрессия ствола мозга, когда она имеется.
Положение миндалин мозжечка у 200 нормальных людей и 25 пациентов с мальформацией Киари 1-го типа приведено в табл. 6-8. Табл. 6-9 показывает изменения, которые возникают, если пределом нижнего положения миндалин считать 2, а не 3 мм.
Табл. 6-8. Расположение миндалин мозжечка от БЗО

* измерения относительно нижней части БЗО
Табл. 6-9. Критерии мальформации Киари 1-го типа

В отличие от выше приведенной информации в качестве нижней границы положения миндалин, пригодной для признания наличия мальформации Киари 1 го типа, часто указывают 5 мм.
Миелография
Ложно негативные результаты наблюдаются только в 6% случаев. КВ должно пройти вплоть до БЗО.
В связи с костными артефактами КТ имеет ограничения в визуализации области БЗО. Надежность исследования повышается при сочетании с интратекально введенным КВ (миелография). Находки: опущение миндалин и/или увеличение желудочков.
Лечение
Показания для хирургического лечения
Основываясь на том, что лучшие результаты наблюдаются в тех случаях, когда пациентов оперируют в течение первых 2 лет после появления симптомов, для симптоматических пациентов рекомендуется раннее хирургическое лечение. Асимптоматичных пациентов можно наблюдать и оперировать позднее, когда у них появятся симптомы. Пациентов, имевших симптомы, не менявшиеся в течение ряда лет, также можно наблюдать. Хирургическое лечение для них показано в случае ухудшения.
Хирургические методики
Наиболее частым видом операций является декомпрессия ЗЧЯ (подзатылочная краниоэктомия) с (или без) другими вмешательствами (обычно комбинируется с пластикой ТМО и шейной ламинэктомией С1 до С2 или С3.
Пациента кладут на живот с валиками под грудной клеткой. Голову фиксируют головодержателем Майфилда. Шею сгибают для увеличения промежутка между затылочной костью и задней дужкой С1. Плечи отводят вниз с помощью липкой ленты. Одно бедро должно быть уложено на валик (мешочек с песком) на случай, если понадобится взять фрагмент широкой фасции для пластики. Разрез делают по средней линии от иниона до ∼ остистого отростка С5. Резецируют кость выше БЗО на ∼3 см вверх и ∼3 см в ширину. ТМО вскрывают Y-образным разрезом и иссекают верхний лоскут. ВНИМАНИЕ: при мальформации Киари поперечные синусы располагаются обычно очень низко (поэтому резекция затылочной кости должна быть небольшой, основной упор должен быть сделан на шейную ламинэктомию и резекцию краев БЗО для того, чтобы устранить компрессию миндалин; компрессия миндалин происходит не в ЗЧЯ). Кроме того, избыточная резекция затылочной кости может привести к выбуханию полушарий мозжечка в образовавшийся дефект, что чревато дополнительными проблемами.
Произведите пластику ТМО надкостницей или фрагментом широкой фасции бедра, чтобы обеспечить достаточно места для миндалин и продолговатого мозга. Некоторые хирурги также дополняют операцию тампонадой верхушки IV-го желудочка мышцей или тефлоном, дренированием сирингомиелитической кисты, если она имеется (фенестрация, обычно через место входа задних корешков, с/или без установкой стента или шунта), шунтированием IV-го желудочка, терминальной вентрикулостомией, вскрытием отверстия Мажанди, если оно закрыто).
Некоторые авторы настоятельно советуют не пытаться удалять спайки между миндалинами, чтобы избежать случайного повреждения жизненно важных структур, включая ЗНМА. Другие же рекомендуют осторожное разделение миндалин и даже их обработку биполярной коагуляцией с тем, чтобы уменьшить их объем.
Некоторые авторы рекомендуют производить трансоральную резекцию ската и зубовидного отростка в случае наличия вентральной компрессии ствола мозга, т.к. они считают, что у этих пациентов может наступить ухудшение после того, как будет произведена только декомпрессия ЗЧЯ. В связи с тем, что было показано, что возникающие при этом изменения являются обратимыми, представляется более целесообразным произвести это вмешательство в случае наступления ухудшения или прогрессирования базиллярной импрессии на МРТ после декомпрессии ЗЧЯ.
Оперативные находки
См. табл. 6-10. Вклинение миндалин имеется во всех случаях (по определению); наиболее часто они расположены на уровне С1 (62%). В 41% случаев имеются сращения между ТМО, арахноидальной оболочкой и миндалинами с закупоркой отверстий Люшка и Мажанди. В 40% случаев отделение миндалин происходит легко.
Табл. 6-10. Оперативные находки при мальформации Киари 1-го типа (71 пациент)

* сосудистые аномалии: ЗНМА была расширена или имела необычное расположение у 8 пациентов (ЗНМА часто спускается до нижнего края миндалин); большие дуральные венозные лакуны
Хирургические осложнения
После подзатылочной краниоэктомии плюс ламинэктомии С1-3 у 71 пациента (с пластикой ТМО у 69 из них) у одного пациента через 36 ч после операции наступила смерть в результате апноэ во время сна. Наиболее частым послеоперационным осложнением было угнетение дыхания (у 10 пациентов), обычно в течение 5 дней, чаще ночью. В связи с этим рекомендуется тщательное наблюдение за дыханием. Другие риски операции: ликворея, вклинение полушарий мозжечка, сосудистые повреждения (ЗНМА и т.д.).
Результаты операций
Пациенты с жалобами на боль обычно хорошо реагируют на операцию. Предоперационная слабость обычно хуже поддается лечению, особенно если уже имеется атрофия мышц. Чувствительность может улучшиться, если задние столбы не поражены, и дефицит обусловлен вовлечением только спинно-таламических трактов.
Ротон считает, что основным эффектом операции является прекращение прогрессирования симптоматики.
Наиболее благоприятные результаты наблюдались у пациентов с мозжечковым синдромом (улучшение у 87% без последующего ухудшения). Факторы, которые коррелировали с плохими исходами: наличие атрофии, атаксия, сколиоз, продолжительность симптоматики более 2 лет.
Табл. 6-11. Отдаленные результаты (69 пациентов, средний срок наблюдения 4 года)

* у этих пациентов было ухудшение до предоперационного уровня (без дальнейшего ухудшения) в течение 2-3 лет после операции; рецидив наступил у 30% пациентов с синдромом компрессии в БЗО и у 21% пациентов с синдромом центрального поражения СМ
Мальформация Киари 2-го типа
Обычно сочетается с миеломенингоцеле (ММЦ) и реже с закрытым расщеплением дужек позвоноков (spina bifida ocсulta).
Патофизиология
Вероятно не связана с фиксацией СМ сопутствующим ММЦ. Более вероятна первичная дисгенезия ствола мозга со множеством других аномалий развития.
Основные находки
Каудальная дислокация цервико-медуллярного сочленения, моста, IV-го желудочка и продолговатого мозга. Миндалины мозжечка расположены на уровне БЗО или ниже его. Вместо обычного изгиба цервико-медуллярного сочленения имеется его деформация в виде перегиба.
Другие возможные находки:
1. клювовидный изгиб четверохолмной пластинки
2. отсутствие прозрачной перегородки с увеличенной межталамической спайкой: считается, что отсутствие прозрачной перегородки обусловлено некрозом с резорбцией в результате ГЦФ, а не врожденного отсутствия
3. слабо миелинизированные мозжечковые листи
4. ГЦФ: имеется в большинстве случаев
5. гетеротопия (патологическая дислокация)
6. гипоплазия фалькса
7. микрогирия
8. дегенерация ядер нижних ЧМН
9. костные аномалии:
A. в области цервико-медуллярного сочленения
B. ассимиляция атланта
C. платибазия
D. базиллярная импрессия
E. деформация Клиппеля-Фейля
10. гидромиелия
11. образование лакун в черепе
Клинические проявления
Проявления связаны с дисфункцией ствола мозга и нижних ЧМН. Манифестация заболевания во взрослом возрасте наблюдается редко. Проявления у новорожденных существенно отличаются от проявлений у детей старшего возраста. Новорожденные более склонны к развитию быстрого неврологического ухудшения со значительной дисфункцией ствола мозга в течение нескольких дней. У более старших детей симптомы развиваются более постепенно и редко проявляются в столь тяжелом виде.
Находки:
1. затруднения глотания (нейрогенная дисфагия) (69%). Проявляется плохим кормлением, цианозом после кормления, назальной регургитацией, продолжительным временем кормления, скоплением слюны. Глоточный рефлекс часто понижен. Более тяжело выражены у новорожденных
2. припадки апноэ (58%): связаны с нарушенной стимуляцией дыхания. Чаще встречается у новорожденных
3. стридор (56%): чаще у новорожденных, обычно хуже на вдохе (при ларингоскопии виден паралич отводящих и иногда приводящих голосовых связок) в связи с парезом Х-го ЧМН. Обычно носит временный характер, но может перейти в остановку дыхания
4. аспирация (40%)
5. слабость верхних конечностей (27%), которая может перейти в тетрапарез
6. опистотонус (18%)
7. нистагм: особенно направленный вниз
8. слабый крик или отсутствие его
9. слабость лицевой мускулатуры
Диагностика
Обзорные краниограммы
Может быть видна цефалофациальная диспропорция в результате врожденной ГЦФ. Образование лакун в черепе (т.н. lukenschadel) наблюдается в 85% случаев (округлые дефекты в черепе с четкими краями, разделенные неравномерно ветвящимися костными полосками). Могут быть низко расположенный внутренний затылочный выступ (укороченная ЗЧЯ), увеличение БЗО (в 70%) и удлинение верхнешейных дужек.
КТ и/или МРТ
Основные находки
A. Z-образная деформация продолговатого мозга*
B. мозжечковая «затычка»
C. четверохолмное сращение (клювовидный изгиб в области четверохолмия)
D. увеличенная промежуточная масса (межталамическое сращение)*
E. удлинение/цервикализация продолговатого мозга
F. низкое прикрепление мозжечкового намета
Сопутствующие находки
A. ГЦФ
B. сирингомиелия в области цервико-медуллярного сочленения (частота по данным до использования МРТ составляет 48-88%)
C. отключенный IV-ый желудочек
D. церебелло-медуллярная компрессия
E. агенезия/дисгенезия мозолистого тела*
* эти признаки лучше оценивать по МРТ
Ларингоскопия
Ее производят у детей со стридором для исключения крупа и других инфекций верхних дыхательных путей.
Лечение
Установите шунт для коррекции ГЦФ (если шунт уже имеется, то проверьте его функционирование)
При нейрогенной дисфагии, стридоре, приступах апноэ рекомендуется ускоренная декомпрессия ЗЧЯ (требуется у 18,7% пациентов с ММЦ); перед проведением рекомендованной декомпрессии всегда необходимо убедиться в том, что у пациента имеется функционирующий шунт!
Хирургическая декомпрессия
NB: плохие оперативные результаты у новорожденных объясняются тем, что многие из неврологических находок могут быть связаны с врожденными (некорректиуемыми) нарушениями, которые не может улучшить декомпрессия. По другой точке зрения, гистологические изменения обусловлены хронической компрессией ствола мозга и сопутствующей ишемией, поэтому ускоренная декомпрессия ствола мозга должна быть произведена как только появляются какие-либо из следующих критических предупреждающих симптомов: нейрогенная дисфагия, стридор, приступы апноэ.
Методика операции: декомпрессия миндалин мозжечка обычно с пластикой ТМО. Пациента укладывают на живот, шея согнута. Производят подзатылочную краниоэктомию и шейную ламинэктомию, которая должна достигать вершины миндалин мозжечка. Между краем БЗО и дужкой С1 обычно удается обнаружить утолщенную дуральную связку, сдавливающую СМ. ТМО вскрывают Y-образным разрезом. При вскрытии ТМО выше уровня БЗО у младенцев требуется осторожность, т.к. они имеют хорошо развитый затылочный синус и могут иметь большие дуральные «озера». Не пытайтесь отделить миндалины от подлежащего продолговатого мозга. В том случае, когда имеется большая сирингомиелитическая полость, делают сиринго-субарахноидальное шунтирование.
Если до операции имеется стридорозное дыхание или парез мышц, отводящих гортань, рекомендуется трахеостомия (обычно временная). Требуется тщательное послеоперационное наблюдение за дыханием (контроль обструкции и снижения стимуляции вентиляции). ИВЛ показана при гипоксии и гиперкарбии.
Исходы
В 68% случаев наступает полное или практически полное разрешение симптомов. В 12% случаев имеется незначительный или умеренный остаточный неврологический дефицит. В 20% случаев улучшения не наступает. В общем, у новорожденных результаты хуже, чем у детей старшего возраста.
Наиболее частой причиной смертности является остановка дыхания (8 из 17 умерших пациентов). Остальные случаи: менингит/вентрикулит (6 пациентов), аспирация (2) и билиарная атрезия (1).
При послеоперационном наблюдении продолжительностью от 7 мес до 6 лет смертность оперированных больных составила 37,8%.
Наиболее важными прогностическими факторами были предоперационное состояние и скорость неврологического ухудшения. Летальность среди младенцев с кардио-пульмональной остановкой, параличом голосовых связок и слабостью рук в течение 2 нед после клинического проявления составила 71%. При более постепенном ухудшении летальность составила 23%. Наихудшим прогностическим фактором в отношении результативности оперативного лечения был двусторонний паралич голосовых связок.
Другие типы мальформации Киари
Мальформация Киари 3-го типа
Встречается редко. Является наиболее тяжелой формой. Смещение структур ЗЧЯ с вклинением мозжечка через БЗО в шейный канал, часто с шейным или подзатылочным энцефаломенингоцеле. Обычно не совместимо с жизнью.
Мальформация Киари 4-го типа
Гипоплазия мозжечка без вклинения мозжечка.
Гринберг. Нейрохирургия
(мальформация Арнольда-Киари) - заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).

Общие сведения
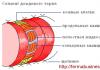
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и др. аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией .
Причины возникновения аномалии Киари
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия , при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация аномалии Киари
Аномалия Киари подразделяется на 4 типа:
Характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией - скоплением цереброспинальной жидкости в центральном канале спинного мозга.
Проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле - врожденной спинномозговой грыжей.
Отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
Заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Симптомы аномалии Киари
Наиболее часто в клинической практике встречается аномалия Киари I типа. Она проявляется ликворногипертензионным, церебеллобульбарным и сирингомиелическим синдромами, а также поражением черепно-мозговых нервов. Обычно аномалия Киари I манифестирует в период полового созревания или уже во взрослом возрасте.
Для ликворногипертензионного синдрома, которым сопровождается аномалия Киари I, характерна головная боль в затылке и шейной области, усиливающаяся во время чихания, кашля, натуживания или напряжения мышц шеи. Может наблюдаться рвота, не зависящая от приема пищи и ее характера. При осмотре пациентов с аномалией Киари выявляется повышенный тонус мышц шеи. Среди мозжечковых нарушений наблюдаются нарушение речи (дизартрия), нистагм , мозжечковая атаксия .
Поражение ствола мозга, расположенных в нем ядер черепно-мозговых нервов и их корешков проявляются снижением остроты зрения, диплопией, расстройством глотания, снижением слуха по типу кохлеарного неврита , системным головокружением с иллюзией вращения окружающих предметов, ушным шумом, синдромом сонных апноэ , повторяющимися кратковременными потерями сознания , ортостатическим коллапсом. Пациенты, у которых имеется аномалия Киари, отмечают усиление головокружения и ушного шума при поворотах головой. Поворот головы у таких больных может спровоцировать обморок. Может отмечаться атрофические изменения половины языка и парез гортани , сопровождающийся осиплостью голоса и затруднением дыхания. Возможен тетрапарез с большим снижением мышечной силы в верхних конечностях, чем в нижних.
В случаях, когда аномалия Киари I сочетается с сирингомиелией, наблюдается сирингомиелический синдром: нарушения чувствительности по диссоциированному типу, онемения, мышечные гипотрофии, тазовые нарушения, нейроартропатии , исчезновение брюшных рефлексов. При этом некоторые авторы указывают на несоответствие размера и местонахождения сирингомиелической кисты распространенности расстройств чувствительности, степени выраженности парезов и мышечной гипотрофии.
Аномалия Киари II и Киари III имеют сходные клинические проявления, которые становятся заметны с первых минут жизни ребенка. Аномалия Киари II сопровождается шумным дыханием (врожденный стридор), периодами кратковременной остановки дыхания, двусторонним нейропатическим парезом гортани , нарушением глотания с забросом жидкой пищи в нос. У новорожденных аномалия Киари II проявляется также нистагмом, повышением мышечного тонуса в верхних конечностях, цианозом кожных покровов, возникающим во время кормления. Двигательные расстройства могут быть выражены в различной степени и прогрессировать вплоть до тетраплегии. Аномалия Киари III имеет более тяжелое течение и зачастую является не совместимым с жизнью нарушением развития плода.
Диагностика аномалии Киари
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ , Эхо-ЭГ , РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. е. гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника , особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение аномалии Киари
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр.) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия . Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций , направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз аномалии Киари
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
… некоторые формы этих пороков вызывают довольно неприятные симптомы, от простого головокружения до инсультов головного и спинного мозга. Симптоматика может долгое время отсутствовать, а затем развиться внезапно, после травмы, гриппа или другой провокации, причем в любом возрасте .Аномалия Арнольда-Киари – это врожденная патология развития ромбовидного мозга , проявляющаяся несоответствием размеров задней черепной ямки и мозговых структур, находящихся в этой области, что приводит к опущению ствола головного мозга и миндалин мозжечка в большое затылочное отверстие и ущемлению их на этом уровне.
Сущность данной аномалии заключается в гетеротопическом расположении мозжечка и продолговатого мозга в расширенном интраспинальном канале. Эта аномалия (мальформация) приводит к развитию сложных краниоспинальных синдромов, которые часто расцениваются неврологами как атипичная форма сирингомиелии и сирингобульбии, рассеянного склероза, а также опухолей задней черепной ямки или спинного мозга. Примерно у 80% пациентов аномалия Арнольда-Киари сочетается с патологией спинного мозга – сирингомиелией, которая характеризуется образованием в спинном мозге кист, вызывающих прогрессирующую миелопатию.
Данное заболевание названо в честь немецкого патологоанатома Юлиуса А. Арнольда (который описал в 1984 году данное заболевание) и австрийского патолога Ханса Киари (который также в 1895 году описал данную аномалию). Частота этого заболевания составляет от 3,3 до 8,2 наблюдений на 100 000 населения. Наиболее распространены аномалии Арнольда-Киари I и II типа; аномалии III и IV типов обычно несовместимы с жизнью:
аномалия Арнольда-Киари I типа
представляет собой опущение структур задней черепной ямки в позвоночный канал ниже плоскости большого затылочного отверстия ( читать
подробнее об аномалии Киари I типа);
аномалия Арнольда-Киари II типа
- происходит каудальная дислокация нижних отделов червя, продолговатого мозга и IV желудочка, нередко развивается гидроцефалия;
аномалия Арнольда-Киари III типа
встречается редко, характеризуется грубым каудальным смещением всех структур задней черепной ямки;
аномалия Арнольда-Киари IV типа
- гипоплазия мозжечка без смещения его вниз.
По данным Н.Н. Яхно и Д.Р. Штульмана (руководство для врачей «Болезни нервной системы» Москва, «Медицина» , 2001) : «… мальформация Киари представляет собой дисгенезию мозжечка в сочетании с широким кругом аномалий ромбовидного, среднео и промежуточного мозга . Выделяют несколько типов мальформации Киари.
Мальформация Киари I (взрослый тип) наиболее частая форма аномалий мозжечка. Этот симптомокомплекс представляет собой одно- или двустороннее опущение миндалин мозжечка через большое затылочное отверстие в позвоночный канал (миндалины - это нижняя часть мозжечка; в норме они расположены выше большого затылочного отверстия). Этому может способствовать каудальное смещение продолговатого мозга. нередко эктопия миндалин мозжечка сочетается с сирингомиелией и аномалиями краниовертебрального перехода. Важнейшим обстоятельством является тот факт, что клинические проявления возникают только на 3-4-м десятилетии жизни. не менее важно, что клинически асимптомная эктопия миндалин мозжечка не требует лечения, являясь случайной находкой на МРТ.
Частота симптомов при аномалии Киари I (по Paul и Jye, 1983 с изменениями) : головная боль – 34%, боль в шее – 13%, опоясывающие боли в руках и/или ногах – 11%, слабость (как минимум в одной из конечностей) – 56%, онемение (как минимум в одной из конечностей) – 52%, потеря болевой и температурной чувствительности – 40%, пошатывание – 40%, горизонтальный и/или вертикальный нистагм – 30%, диплопия – 13%, дисфагия – 8%, рвота – 5%, дизартрия – 4%, головокружение – 3%, глухота – 3%, обмороки – 2%, онемение лица – 3%.
Как и при других процессах, поражающих цервико-медуллярный переход, при аномалии Киари I нередко наблюдается «нистагм, бьющий вниз». Наличие нарастающих очаговых симптомов (мозжечковых, стволовых, спинальных), а также гидроцефалии – повод к обсуждению показаний субокципитальной декомпрессии. В подобной ситуации необходим сугубо индивидуальный подход, чтобы избежать как неоправданного вторжения, так и промеделния с хирургической коррекцией. Операция приводит к выздоровлению или улучшению у 2/3 больных. Благоприятным прогностическим признаком служит наличие только мозжечковых симптомов и дислокация мозжечка не ниже СI позвонка. Возможны рецидивы болезни на протяжении 3 лет после операции.
Мальформация Киари II (детский тип) складывается и смещения мозжечка, ствола и IV желудочка через большое затылочное отверстие. Характернейший признак – сочетание с менингомиелоцеле в поясничной области. Неврологические дефекты проявляются на фоне аномалий затылочной кости и шейного отдела позвоночника. Всегда имеется гидроцефалия, часто – стеноз водопровода мозга. Неврологические симптомы имеются уже при рождении. Менингомиелоцеле требует операции в первые дни жизни. Последующая субокципитальная декомпрессия может привести к значительному улучшению. Большинство больных нуждаются в шунтирующей операции, особенно если имеется стеноз водопровода мозга.
Мальформация Киари III. Менингоэнцефалоцеле в нижней затылочной или верхней шейной области в сочетании с пороками развития нижней части ствола мозга, основания черепа и верхних шейных позвонков. Мозговая грыжа включает мозжечок и в половине случаев – затылочную долю. Очень редкий симптомокомплекс с плохим прогнозом даже при своевременном хирургическом вмешательстве.
Мальформация Киари IV. аномалия представлена изолированной гипоплазией мозжечка и не является симптомокомплексом Киари в современном понимании.»
Современная патоморфология выделяет три основные типа этой аномалии : I - проникновение миндалин мозжечка в шейный отдел позвоночного канала; II - вклинение дисплазированного мозжечка в большое затылочное отверстие в сочетании с удлинением ствола мозга; III - изолированное тотальное смещение структур заднего мозга в расширенное затылочное отверстие, сопровождаемое образованием грыжи. I тип обычно не сопровождается поражением спинного мозга и выявляется чаще у взрослых при помощи КТ и ЯМР. По данным аутопсии у детей с менингомиелоцеле аномалию Арнольда-Киари II типа обнаруживают в 95-100% случаев.
До настоящего времени патогенез патологии окончательно не установлен . По всей вероятности, этих патогенетических факторов три: (1) первый - наследственно обусловленные врожденные остеоневропатии, (2) второй - травматические повреждения клиновидно-решетчатой и клиновидно-затылочной части ската вследствие родовой травмы, (3) третий - гидродинамический удар ликвора в стенки центрального канала спинного мозга.
При этой врожденной аномалии миндалины мозжечка имеют вид пленок, покрывающих дорсальные отделы продолговатого и спинного мозга. Дистальные отделы продолговатого мозга опускаются вниз и располагаются в позвоночном канале на уровне верхних шейных позвонков. В связи с опущением продолговатого мозга опускаются и нижние отделы IV желудочка. Следовательно, отверстие Мажанди при этом оказывается на уровне большого затылочного отверстия или еще ниже, а корешки каудальной группы черепно-мозговых нервов и верхних шейных сегментов спинного мозга в различной степени вытягиваются, что обуславливает компрессию каудального отдела продолговатого мозга, миндалин мозжечка и краниальных отделов спинного мозга.
Помимо непосредственно компрессионного фактора при аномалии Арнольда-Киари имеет значение нарушение ликвородинамики . В норме спинномозговая жидкость (ликвор) свободно циркулирует в субарахноидальных пространствах головного и спинного мозга. На уровне большого затылочного отверстия субарахноидальные пространства головного и спинного мозга соединяются, что обеспечивает свободный отток ликвора от головного мозга. При аномалии Арнольда-Киари низко расположенные миндалины мозжечка затрудняют свободную циркуляцию спинномозговой жидкости между головным и спинным мозгом. Миндалины блокируют большое затылочное отверстие, как «пробка затыкает бутылочное горлышко». В результате нарушается отток ликвора и развивается гидроцефалия.
Клинические симптомы постепенно и медленно прогрессируют в течение 3-6 лет и характеризуются вовлечением в процесс верхнешейного отдела спинного мозга и дистального отдела продолговатого мозга с нарушением функции каудальной группы черепно-мозговых нервов, а также мозжечка.
Исходя из вышеизложенного, различают три неврологических синдрома :
(1 ) При церебеллобульбарном синдроме клинически устанавливается нарушение тройничного (V), лицевого (VII) и вестибулокохлеарного (VIII) нервов, нервов вагусной системы и подъязычного нерва (IX-XII), спонтанный нистагм, бьющий вниз, вестибулокохлеарные нарушения, головокружения, нарушения дыхания, носовой оттенок речи, поперхивания, нарушения глотания, односторонний парез мягкого неба, снижение глоточного рефлекса, охриплый голос, статическая и динамическая атаксия, нарушение координации, парез ног (спастического характера).
Рентгенологически и при МРТ на боковом снимке черепа имеется расширение сагиттального размера позвоночного канала на уровне СI и СII, при каротидной ангиографии огибание снизу миндалины мозжечка задненижней мозжечковой артерией.
(2 ) При сирингомиелитическом синдроме клинически отмечаются охриплость голоса, нарушение акта глотании, нистагм, атрофия мышц языка, анестезия в некоторых зонах лица, слабость в конечностях с повышением сухожильных рефлексов и мышечного тонуса по спастическому типу, сегментарно диссоциированные расстройства чувствительности на уровне верхнешейных сегментов спинного мозга.
Рентгенологически на боковом снимке визуализируется гипоплазия задней дужки атланта, гипоплазия эпистрофея, деформация и сужение большого затылочного отверстия, неравномерное развитие или недоразвитие боковых масс атланта, расширение интраспинального канала на уровне СI и СII. В этих случаях показана МРТ или инвазивные рентгенологические методы исследования.
(3 ) При пирамидном синдроме клинически отмечается легкий спастический тетрапарез со снижением мышечной силы в руках и ногах. Сухожильные и периостальные рефлексы на конечностях повышены, брюшные рефлексы снижены или не вызываются.
Рентгенологически определяются сопутствующие признаки нарушения развития костных элементов краниовертебральной зоны такие, как гипоплазия атланта, недоразвитие зубовидного отростка аксиса, укорочение атланто-затылочного промежутка. Верхушка зуба распологается выше линии Мак Грегора, увеличен сагиттальный размер большого затылочного отверстия и интраспинального канала на уровне СI и СII.
Типичная клиническая картина аномалии Арнольда-Киари характеризуется следующими симптомами : боль в шейно-затылочной области усиливающаяся при кашле, чихании, снижение болевой и температурной чувствительности в верхних конечностях, снижение мышечной силы в верхних конечностях, спастичность верхних и нижних конечностей, обмороки, головокружения, снижение остроты зрения, в более запущенных случаях присоединяются: эпизоды апноэ (короткая остановка дыхания), ослабление глоточного рефлекса, непроизвольные быстрые движения глаз.
Любопытным клиническим феноменом у этих больных является провокация симптомов (боли в шее, в затылке, парестезии, онемение половины тела, приступы внезапного падения, обморок) кашлем, чиханием, смехом, натуживанием, пробами Вальсальвы, Нери, Дежерина, что обусловлено динамическим усилением компрессии цервикомедуллярных структур в момент внезапного повышения внутричерепного давления с ущемлением миндалин мозжечка в большом затылочном отверстии.
Клиническая манифестация аномалии Арнольда-Киари подчас у взрослых и даже пожилых людей постоянно дает повод к диагностике опухоли задней черепной ямки или верхних отделов спинного мозга. Правильному распознаванию способствуют иногда имеющиеся у больных внешние признаки дизрафии (короткая шея, низкая линия оволосения на шее), а также обнаружение на рентгенограммax (а также на КТ/МРТ) кранио-спинальных признаков костных аномалий.
В настоящий момент методом выбора при диагностике аномалии Арнольда-Киари является МРТ головного мозга шейного и грудного отделов спинного мозга (для исключения сирингомиелии).
Возможна уьтразвуковая диагностика аномалии Арнольда-Киари у плода . На современном этапе к возможным эхографическим признакам аномалии Арнольда-Киари относят внутреннюю гидроцефалию, а также типичную форму мозгового черепа типа «лимон» и изображение мозжечка в виде «банана». Однако по мнению некоторых исследователей эти признаки не отличаются высокой специфичностью по отношению к пороку Арнольда-Киари. Для решения этой проблемы показано использование не только горизонтальной, но и других плоскостей сканирования (фронтальной, сагиттальной), что позволяет выявить ряд высокоинформативных критериев аномалии Арнольда-Киари у плода. Частота эхографической выявляемости таких признаков, как отсутствие большой цистерны и удлинение ножек мозга, убедительно показала преимущества сагиттальной плоскости сканирования мозга для диагностики основного макроскопического признака порока Арнольда-Киари - вклинения частей мозжечка в большое затылочное отверстие. Получение изображения головного мозга плода в сагиттальной плоскости во II триместре беременности не является сложной технической задачей. В связи с этим использование сагиттальной плоскости сканирования мозга является одной из наиболее информативных плоскостей для диагностики или исключения аномалии Арнольда-Киари у плода как во II, так и в III триместрах беременности.
Обнаружение признаков аномалии Арнольда-Киари у плода может явиться показанием к исключению дефектов развития позвоночника (включая инвазивные), при этом даже полное отсутствие ультразвуковой информации о состоянии позвоночника указывает на наличие Spina bifida более чем в 95% случаев.
Лечение . При «асимптомном варианте» - динамическое наблюдение с ежегодным проведением обследования. Если единственным симптомом заболевания является незначительной интенсивности болевой синдром, для лечения применяется консервативная терапия, которая включает в себя различные схемы с примененим нестероидных противовоспалительных препаратов и миорелаксантов. Периодически проводят дегидратационную терапию. При отсутствии эффекта от консервативной терапии в течение 2-3 месяцев или наличии у пациента неврологического дефицита (онемение, слабость а конечностях и т.д.) показано проведение операции (ламинэктомия, расширение затылочного отверстия и др.). В части случаев во время хирургической ревизии и устанавливается окончательный диагноз. Целью операции является устранение сдавления нервных структур и нормализация тока цереброспинальной жидкости, для чего производится увеличение объема задней черепной ямки. В результате лечения, как правило, уменьшается или исчезает головная боль, частично восстанавливаются чувствительность и двигательные функции.
Аномалия Арнольда-Киари I-II встречается достаточно часто.
Аномалия Арнольда-Киари I представляет собой тонзиллярную эктопию, при которой деформированная миндалина мозжечка смещается ниже уровня большого затылочного отверстия в верхнюю часть цервикального канала (рис. 3.1). Тонзиллярная эктопия на 3-4 мм является бессимптомной. При смещении миндалин мозжечка на 5 мм и более появляются начальные клинические признаки. Эта аномалия развития в 20-25% наблюдений связана с сирингогидромиелией и умеренной гидроцефалией. Часто она сочетается с аномалией краниовертебральной области: базилярной импрессией (25% случаев), сращением с затылочной костью (10%), синдромом Клиппеля-Фейля (10%), незавершенной оссификацией С, (5%) (рис. 3.2).
Применение МРТ предпочтительнее, чем КТ. При МРТ на срединном сагиттальном срезе, лучше наТ1 ВИ, определяются удлиненные миндалины мозжечка, располагающиеся ниже плоскости большого затылочного отверстия. Пролапс должен быть не менее 3 мм. Большая затылочная цистерна мала или отсутствует. Часто определяются также смещение ствола мозга кпереди и сглаженность моста. Смещение ствола мозга приводит к появлению симптома «ступеньки» в месте перехода продолговатого мозга в спинной.
 а
а
Рис. 3.1. Аномалия Арнольда-Киари I. MPT.
а - корональная плоскость, Т1-ВИ. Миндалины мозжечка удлинены, располагаются ниже плоскости большого затылочного отверстия.
б - сагиттальная плоскость, Т1-ВИ. Большая затылочная цистерна отсутствует. Ствол мозга смещен кпереди, отмечается сглаженность моста.

Рис 3.2. Аномалия Арнольда-Киари,
сочетающаяся с сирингогидромиелией.
а - схема срединного сагиттального изображения. Низкое расположение миндалины мозжечка (большая стрелка), сирингомиелия (стрелки).
б - МРТ, Т1-ВИ, сагиттальная плоскость. Миндалины мозжечка пролабируют ниже уровня большого затылочного отверстия. Большая цистерна мозга не прослеживается. Продолговатый мозг смещен кпереди, понтомедуллярная цистерна сужена. Сирингомиелия на уровне C 2 -Th r На уровне Th 2 прослеживается расширение центрального спинномозгового канала.
в - МРТ, Т2-ВИ, аксиальный срез на уровне большого затылочного отверстия. Миндалины мозжечка плотно выполняют затылочное отверстие, субарахноидальное пространство не прослеживается.
Аномалия Арнольда - Киари II - комплекс пороков, поражающих позвоночник, череп, твердую мозговую оболочку, ромбовидный мозг. В отличие от аномалии Арнольда - Киари I, она всегда связана с некоторыми формами спиналь-ного дизморфизма, а также менингоцеле или миеломенингоцеле и гидроцефалией (рис. 3.3). Достаточно часто сочетается с супратенториальными пороками.
|
|


Рис. 3.3. Беременность 32-33 недели.
Аномалия Арнольда-Киари II. Вентрикуломегалия. МРТ. Т2-ВИ.
а, б, в - изображения головного мозга в аксиальной, сагиттальной и корональной плоскостях. Выраженное расширение задних отделов боковых желудочков. Низкое расположение миндалин мозжечка. Ангуляция шейного отдела позвоночника. Спинномозговая грыжа крестцового отдела позвоночника.
Пороки при аномалии Арнольда-Киари II многочисленные и сложные. Лучевые исследования помогают различать их степени и комбинации.
Аномалии Арнольда-Киари II может сочетаться с другими пороками развития (черепа, позвоночника, твердой мозговой оболочки, желудочков мозга, аномалиями ствола мозга, спинного мозга, мальформациями кортикального развития).
Дисплазия губчатых костей, ведущая к фестончатому истончению свода черепа, является не мальформацией, а только сопутствующим симптомом и редко встречается у детей старше 6 месяцев. Она проявляется низким расположением синусного стока и поперечных синусов, следствием чего является маленькая и мелкая задняя черепная ямка; расширенным большим затылочным отверстием, тенториальной гипоплазией с широкой вырезкой; червь и мозжечок могут пролабировать вверх через тенториальную вырезку (псевдоопухоль мозжечка). Также могут наблюдаться различной степени гипоплазия серпа большого мозга или сращение его с примыкающей извилиной, что придает межполушарной щели фестончатый или губчатый вид, уплотнение костей. Скат может казаться вогнутым в задних отделах.
Дизгенезия ствола мозга ведет к смещению вниз продолговатого мозга и мозжечка. Происходит изгиб продолговатого мозга, выпячивание покрышки и переднемедиальный рост мозжечка по бокам ствола мозга.
Размеры боковых желудочков варьируют от нормальных до значительно дилатированных, часто асимметричных, с выступающими височными рогами, передние и задние рога могут быть отшнурованы. Таким образом, желудочки часто имеют фестончатый вид.
Прозрачная перегородка либо отсутствует, либо сообщается с желудочковой системой.
III желудочек часто увеличен, с деформированным передним карманом. IV желудочек нередко удлинен, имеет небольшие размеры, смещен каудально.
Может определяться стеноз или окклюзия водопровода мозга.
Аномалия Арнольда-Киари II может сочетаться с мальформациями кортикального развития. Наиболее частыми ассоциированными кортикальными дисплазиями являются гетеротопия серого вещества, полимикрогирия. Также к сочетанным аномалиям относят аномалии полосатого тела (обычно частичное отсутствие либо гипоплазия).
Спинальный дисрафизм, миеломенингоцелле присутствуют почти всегда. Наиболее часто встречающейся патологией является миеломенингоцелле. В 75% случаев поражается пояснично-крестцовый отдел позвоночника. В спинном мозге может определяться сирингогидромиелия. Поражение спинного мозга в ряде случаев обусловлено сдавлением липомой, диастематомиелией.
Аномалия Арнольда-Киари III встречается редко и представляет собой аномалию Арнольда-Киари IIс низким затылочным и высоким шейным энцефалоцелле.
Аномалия Арнольда-Киари IV характеризуется выраженной гипоплазией мозжечка. Это очень редкая аномалия, не существующая как отдельная нозологическая единица. Определяются отсутствие или гипоплазия мозжечка, маленький ствол мозга, большая задняя черепная ямка.