Данная информация предназначена для специалистов в области здравоохранения и фармацевтики. Пациенты не должны использовать эту информацию в качестве медицинских советов или рекомендаций.
Т-клеточные иммунодефициты
Первичные Т-клеточные дефициты - это редкие наследственные нарушения, поражающие развитие и функцию Т-клеток. Эти нарушения обычно проявляются в грудном или раннем детском возрасте; однако, возраст появления симптомов может варьировать в зависимости от в основе лежащего генного дефекта. Хотя Т-клеточные иммунные реакции могут быть нарушены селективно, тем не менее, с этим часто ассоциируют ненормальные В-клеточные функции, отчасти из-за сопутствующих внутренних В-клеточных дефектов, но также и потому, что выработка большинства антител зависит от помощи Т-клеток.
ТЯЖЕЛЫЕ КОМБИНИРОВАННЫЕ СИНДРОМЫ ИММУНОДЕФИЦИТА
Тяжелые комбинированный иммунодефицитный синдром (SCID) - это наследственное нарушение у детей, характеризующееся глубоко дефективной или отсутствием Т клеточной и В клеточной функций. SCID часто оказывается фатальным в течение первого года жизни, несмотря на проведение терапевтической трансплантации стволовых клеток или в случае дефицита аденозин деаминазы (ADA) про-веденного замещения фермента. Ранее выявление пораженных пациентов до развития у них оппорту-нистических инфекций является критическим для достижении благоприятного исхода. Диагноз SCID считается подтвержденным, когда у пораженного ребенка определяется лимфопения (Несмотря на имеющуюся генетическую гетерогенность, у пациентов с SCID имеется много общего в течение первых 6 месяцев жизни.
Обычно у пораженных детей могут развиться следующие инфекции:
Бактериальные
Грам-негативный сепсис
Диссеминация бациллы Calmette-Guerin после иммунизации
Вызванный грибками и простейшими
Кандидиаз
Аспергиллез
Pneumocystis carinii пнемовния
Вирусные
Цитомегаловирус
Вирусы Parainfluenza
Аденовирусы
Респираторный синцитиальный вирус
Диссеминированная варицелла (ветряная оспа)
Вакцинально-приобретенный паралитический полимиелит
Molluscum contagiosum
Также может иметь место отсутствие набора веса вторичное к диарее или мальабсорбции. Появление раннего начала эритематозной макулопапулярной сыпи, не реагирующей на медицинское лечение, может указывать на хроническое заболевание трансплантант против хозяина (GVHD) при пересадке материнских Т клеток. У большинства пациентов с SCID имеется гипоплазия тимуса и отсутствие или мелкие, слаборазвитые лимфатические узлы и тонзиллы; гепатоспленомегалия может выявляться у детей пораженных материнской GVHD. Рентгенограммы грудной клетки часто дают отсутствие тени тимуса и слабо выраженный легочной рисунок, несмотря на наличие существенных респираторных симптомов.
У большинства SCID пациентов количество периферических CD3+ Т -клеток составляет 500 cells/mm3 или менее (нормальные границы 3000-6500 cells/mm3) и различное количество В и естественных киллеров (NK) лимфоцитов в зависимости от основного генетического дефекта. SCID можно класси-фицировать в соответствие с наличием или отсутсвтием В и NK клеток на T-B+NK+ , T-B+ NK- , T- B-NK+ , T-B-NK- и атипичный Т+В+ синдромы (табл. 1). У пациентов с ADA дефицитом имеется наи-меньшее количество циркулирующих лимфоцитов, в то время, как у пациентов с неизвестным аутосо-мально рецессивным (AR) T+B+ SCID, ZAP-70 дефицитом и материнским Т-клеточным внедрением имеется наибольшее количество лимфоцитов, часто в пределах нормальных границ. Пациенты с SCID анергичны в отношении кожного теста на гиперчувствительность отсроченного типа и результа-ты измерения in vitro Т-клеточной функции оказываются существенного сниженными до 10% или ме-нее от нормальных значений.
Кроме малого количества циркулирующих Т-лимфоцитов, выраженной гипогаммаглобулинемии отме-чается и особенное снижение IgG. Уровень IgG сыворотки в нормальных границах обычно является отражением материнских антител у маленьких детей с SCID или внутривенного введения гаммаглобу-лина (IVIG). Уровни IgA и IgM в сыворотке ранжируются от их отсутствия до нормальных возрастных значений. Наличие определяемого сыворотчного IgE и эозинофилии обычно имеет место у детей с материнским GVHD или синдромом Omenn (OS). Несмотря на наличие сывороточных иммуноглобу-линов у некоторых SCID пациентов выработка антиген-специфических антител отсутствует; поэтому использование антитело-зависимых методов, таких как энзим-связанный иммуносорбентный тест для скрининга экспозиции к инфекционным агентам у пациентов с SCID дают фальшь-негативные резуль-таты или оказываются фальшь-позитивными из-за введения IVIG. Вместо этого следует применять непосредственное определение антигена иммунофлюоресценцией или путем реакции полимеризации цепи для диагностики инфекции у иммунодефитных пациентов.
Единственным лечением SCID является реконституция гематопоетической стволовой клетки. Опти-мальным лечением является трансплантация костного мозга (BMT) или трансплантация перифериче-ских стволовых клеток от тканесовместимого брата или сестры. К сожалению, для большинства паци-ентов HLA-идентичный семейный донор отсутствует. Часто проводится Т-клеточно-опустошенная гаптоидентичная BMT от родителей и оказывается успешной для многих пациентов с SCID. Также для реконституции иммунной системы применялась matched unrelated BMT или трансплантация стволо-вой клетки крови пупочного канатика. Основными осложнениями трансплантации являются отторже-ние трансплантанта, GVHD, инфекции и токсичность химиотерапии. Некоторым пациентам требуется продолжительная иммуносупрессия для контроля GVHD или пожизненный IVIG, если приживление донорских В клеток не удалось осуществить, а нормальная В-клеточная функция не восстановилась.
Х-связанный тяжелый комбинированный синдром иммунодефицита
На Х-связанный SCID (ХSCID) приходится от 30 до 40% всех случаев SCID и, как
считается, он развивается у 1-2 на 100 000 родов. Его тип наследования был
установлен на основании большого числа родословных, где мальчики из последующих
поколений умерли в раннем детстве в результате не-управляемых вирусных или
грибковых инфекций. Дефект при ХSCID был идентифицирован в 1993 го-ду, как
обычная g цепь (gс) рецептора интерлейкина (IL)-2. Последующие исследования
показали, что gс также является частью рецепторов для четырех дополнительных
цитокинов: IL-4, IL-7, IL-9 и IL-15. Молекула gс кажется незаменимой для
внутриклеточной трансмиссии сигналов активации возни-кающих при цитокин-цитокин
рецепторном взаимодействиях, которые необходимы для пролиферации и созревания
лимфоцитов. Отсутствие gс-содержащих рецепторных комплексов проявляется ранней
приостановкой Т-клеточного и NK-клеточного развития и выработкой незрелых
В-клеток, которые вы-зывают дефективное переключение изотипов необходимое для
выработки IgG, IgA IgE. Взаимодей-ствие между IL-7 и его рецептором является
особенно критическим для дифференциации лимфоид-комиттированных клеток
предшественников из плюрипотентных стволовых клеток. Этот иммунодефи-цит
представляется прототипичную T-B+NK- форму SCID.
Большинство пациентов мужского пола пораженных XSCID имеют абсолютное количество
лимфоци-тов менее, чем 2000 cells/mm3 в периферической крови, с менее чем 200
cells/mm3 CD3+ T клеток (границы 0-800 клеток/мм3), менее, чем 100 cells/mm3 NK
клеток и повышенный процент (часто >75%) В лимфоцитов (табл.2). Уровни IgG и IgA
в сыворотке чрезвычайно низки и специфическая выработка антител отсутствует. И
наоборот, уровни IgM и IgE в сыворотке могут оказаться нормальными в ре-зультате
материнского Т-клеточного внедрения. In vitro Т-клеточная и NK-клеточная функции
оказы-ваются, как правило, слабыми. У большинства детей пораженных XSCID
присутствуют определяемые при HLA типировании материнские Т лимфоциты в их
крови. Явный GVHD может развиться, если присутствует значительное количество
материнских Т клеток, способных реагировать на полученные от родителей HLA
антигены. Наличие лимфоцитоза, нормальные уровни IgM и IgE, гепатомегалия,
лимфаденопатия и хронической сыпи в результате материнской GVHD часто
задерживают выявление XSCID.
Множественные различные мутации в гене IL2RG были идентифицированы у XSCID
потомков. В от-личие от кистозного фиброза ни одной обычной мутации не
происходит при XSCID; тем не менее, не-которые «hot spots» были идентифицированы
в гене, в котором часто идентифицируются мутации. Как было показано, некоторые
мутации оказывают относительно слабые воздействия на функцию gс; у таких
пациентов имеется тенденция к меньшей лимфопении, лучше сохраняемой Т-клеточной
функ-ции и более близким к норме уровням в сыворотке Ig. Эти дети часто
классифицируются, как имею-щие неизвестный AR T+ B+ SCID, задерживающий
распознание их в основе лежащего генетического дефекта. Так как в большинстве
XSCID семей имеются отдельные мутации, то последовательные анализы кодирующих
регионов гена IL2RG должны производиться для характеристики вредных мута-ций.
Секвенсирование материнской ДНК также может осуществляться для подтверждения
мутаций; однако, у более, чем 50% пациентов мужского пола с XSCID имеется
спонтанно возникшая IL2RG му-тация без доказательства наследственной мутантной
материнской Х хромосомы при секвенировании ДНК или при не слепом характере
инактивации Х хромосомы. Гистосовместимый или гаплоидентич-ный ВМК или
трансплантация периферической или стволовой клетки пупочного канатика дают
иммунную реконституцию у большинства мальчиков с XSCID. In utero ВМТ успешно
лечит пораженные пло-ды, а генная терапия кажется должна стать реальной
возможностью
JAK3 энзимный дефицит
У девочек и некоторых мальчиков с типичным T-B+NK- SCID фенотипом отсутствуют мутации в IL2RG гене. Эти пациенты, многие из которых рождены от родителей родственников, вместо этого имеют AR SCID вызванный мутациями в обеих аллелях JAK3 энзима. Дефицит JAK3 энзима был впервые опи-сан в 1995 году и на него может приходиться от 10 до 20% всех случаев SCID. JAK3 - это цитоплазма-тическая тирозин киназа, которая связана с gс и необходима для трансдукции цитокин-связанных сигналов с gс-содержащих цитокиновых рецепторов. Мутации в JAK3 препятствуют прохождению сигнала через эти рецепторы, подтверждая тем самым наблюдение, что gс функция находится в абсолютной зависимости от активации JAK3, располагающихся ниже по току прохождения сигнала. JAK3 мутации различны и имеют тенденцию быть уникальными для каждого отдельного семейства. Многие исклю-чают JAK3 mRNA или протеиновую экспрессию, но описаны исключения, которые проявляются более слабо выраженным фенотипом SCID и выработкой некоторых Т и NK клеток.
Дефицит интерлейкин-7 рецептора
Дефекты цепи a IL-7-рецептора (IL-7Ra) представляют собой редкие причины AR SCID. Фенотип IL-7Ra дефицита схож с таковым XSCID и JAK3 дефицитом при существенном исключении. В отличие от этих иммунных дефектов IL-7Ra дефицит не приостанавливает развитие NK-клеток и рассматрива-ется как T- B- NK+ форма SCID.
Дефицит интерлейкина-2
Были опубликованы сообщения об отдельных семьях с дефектами в выработке IL-2. У пациентов с IL-2 дефицитом сохраняются относительно нормальные количества периферических лимфоцитов (T+ B+ SCID) и гипогаммальбуминемия. In vitro функция Т-клеток снижена, но поддается коррективроке при добавлении рекомбинанта IL-2. В основе лежащие генетические дефекты у этих пациентов неизвестны, но как считается, они поражают регуляцию транскрипции гена IL-2. Парентеральная терапия ре-комбинантом IL-2 может дать частичную иммунную реконституцию у этих пациентов.
Дефицит RAG 1 и 2
Дети с дефектами лимфоцито-специфическими рекомбиназа-активирующими генами (RAG)
1 или 2 (RAG1 и RAG2) были впервые описаны в 1996 году. RAG 1 или RAG 2
проявляются первичной фор-мой T-B-NK+ SCID и на них может приходиться от 10 до
20% всех случаев. Функция RAG 1 и RAG 2 незаменима для поколений Т-клеточных и
В-клеточных антигенных рецепторов (TCR и BCR, соответ-ственно). Во время
Т-клеточного и В-клеточного онтогенеза хромосомальная ДНК содержащая раз-личные
вариабельные (V), диверсивные (D) и добавочные (J) сегменты TCR генов
иммуноглобулина перестраивается для выработки функциональных антигенных
рецепторов. V(D)J рекомбинация дает диверсиновсть антигенного рецептора и
способность иммунной системе человека реагировать на бо-лее, чем 108 антигенов.
Неспособность совершать V(D)J рекомбинацию проявляется в остановке со-зревания
Т-клеток и В-клеток на ранней стадии лимфоцитарной дифференцировки при полном
отсут-ствии всех Т и В клеток и агаммаглобулинемией. NK клетки не экспрессируют
антиген-специфических рецепторов и их развитие не нарушается при дефективной
RAG-1 или RAG-2 функции. Мутации как в RAG1, так и в RAG2 были идентифицированы
у потомком лиц с T-B-NK+ SCID. Тяжелые RAG1 мута-ции развиваются во внутренних
фрагментах протеина и могут оказаться более частыми, чем отклоне-ния в RAG2.
Синдром Omenn’a (OS)
OS - это редкое AR нарушение, описанное в 1965 году Omenn, как SCID,
характеризующийся следующими симптомами:
Физикальные данные
Эритродерма
Лимфаденопатия
Гепатоспленомегалия
Неспособность набора веса вторичная к диарее
Генерализированный отек
Лихорадка
Лабораторные данные
Гипоальбуминемия
Эозинофилия (>1000 клеток на мм3)
Изменяющееся число лимфоцитов
От сниженного до повышенного количество CD3+ Т клеток
Отсутствие В клеток
Нормальное количество NK клеток
Явно дефективная Т-клеточная и В-клеточная функции
Гипогаммаглобулинемия
Существенного пониженные уровни IgG, IgM и IgA
Гипер-IgE (>1000 IU/mL)
В 1998 году у пациентов с OS были идентифицированы мутации в RAG1 или RAG2,
проявлявшиеся в частичной V(D)J активности рекомбиназы и развитием редко
активированных, но анергичных, олиго-клональных Т клеток.
Эти клинические проявления характерны для OS и отличают его от других форм SCID.
В течение многих лет считалось, что OS является тяжелым вариантом материнского
GVHD, но внедренных ма-теринских Т лимфоцитов определить не удалось. У
пораженных детей количество периферических Т лимфоцитов оказывалось от
существенного сниженного до нормального уровней. Во многих случаях количество
CD3+ Т лимфоцитов оказывалось нормальным, но отсутствовали циркулирующие В
клет-ки. Иммуноглобулины сыворотки не определялись, за исключением существенного
повышенного уров-ня IgE (часто >1000 IU/mL). Также как при полном дефиците RAG 1
или 2 дефиците, количество NK клеток остается нормальным у пациентов с OS. OS
рассматривается как T- B- NK+ форма SCID, но на-личие олигоклональных Т клеток,
которые развиваются благодаря редким успешным V(D)J рекомби-национным
проявлениям, путает диагноз. Гипер IgE обусловлен наличием в не-лимфоидных
тканях редких В клеток, которые простимулированы на выработку IgE клетками
хелперами, направленными на выработку IL-4 IL-5 и обычно встречающихся при
гиперчувствительности или аллергических забо-леваниях. Наличие лимфаденопатии
отличает OS от других форм SCID без материнской GVHD. Гис-тологическое
исследование лимфатических узлов при OS выявляет нарушение архитектуры,
отсутст-вие образования фолликул и чрезмерную инфильтрацию эозинофилами,
гистиоцитами и активиро-ванными Т клетками. Кожа также массивно инфильтрирована
воспалительными клетками.
Анализ секвенции ДНК у пациентов с OS обнаруживает мутации в RAG1 или RAG2,
которые не пол-ностью устраняют V(D)J активность рекомбиназы. По неизвестным
причинам частичная RAG1 или RAG2 функция проявляется в более продуктивной
перестройке TCR, чем BCR, так олигоклональные Т клетки присутствуют, а
циркулирующие В клетки очень редки. Интересно, что большинство поражен-ных детей
рождены от родителей не имеющих родственных связей. До ВМТ, необходимо исключить
материнскую GVHD. Эти дети также часто оказываются тяжело больными с лихорадкой,
протеин-теряющей энтеропатией и генерализованным отеком из-за воспаления
кишечника и кожи. Аблативная хемотерапия и иммуносупрессия необходимы для
предотвращения отторжения трансплантанта акти-вированными аутологичными Т
лимфоцитами. Предпочтение отдается ткане-совместимой ВМТ, так у пациентов с OS
повышен риск неудачи гаплоидентичной трасплантации.
Тяжелый комбинированный Navajo синдром
Navajo SCID - это AR мутация случающаяся приблизительно у 1 из 2000 живых
новорожденных у Ath-abascan Native Americans. В основе лежащий генетический
дефект остается неизвестным но маппиру-ется на хромосоме 10р при проведении
анализа потомков. Клинические проявления Navajo SCID сходны с таковыми при
других формах SCID за исключением необычного явления наблюдаемого у большинства
пациентов в течение первых 4 месяцев жизни - не связанного с вирусом герпеса
ораль-ных и генитальных язв. Пораженные дети дают лимфопению (300-1800 клеток на
мм3), при количест-ве Т и В клеток менее, чем 200 клеток на мм3. In vitro
функция Т клеток и уровни сывороточного имму-ноглобулина существенно снижены, в
то время, как NK клетки имеются в избытке и их активность нормальна, поэтому
Navajo SCID класссифицируется как T- B- NK+ SCID, но не связанный с дефектив-ной
RAG-1 или RAG-2 функцией. Наличие NK активности осложняет проведение ВМТ
увеличивая риск отторжения трансплантанта. Интенсивное пре-ВМТ аблативное
кондиционирование необходимо для осуществления трансплантации у пораженных
детей.
Дефицит аденозин деаминазы
На дефицит ADA приходится приблизительно 15% всех случаев SCID. Впервые он был
описан в 1972 году. В отличие от других форм SCID, при которых мутации
специфически поражают Т- клеточные и часто В- клеточные функции, ADA дефицит
проявляется метаболическим отравлением всех клеток, с наиболее выраженными
воздействиями на лимфоциты и лимфоидные прогениторы. ADA дефицит - это наиболее
частая форма T- B- NK- SCID.
ADA - это широко распространенный энзим пути расщепления пуринов, который
катализирует деами-нацию аденозина и деоксиаденозина в инозин и в деоксиинозин.
Отсутствие ADA проявляется накоп-лением этих субстратов интрацеллюлярной и в
экстрацеллярной жидкостях. Значительно повышен-ный уровень деоксиаденозин
трифосфата в лимфоцитах связан с интрацеллюлярным захватом и фосфориляцией
деоксиаденозина, что может объяснить почему лимфоциты так чувствительны к
ток-сичным побочным эффектам этих метаболитов. Аденозин, деоксиаденозин и
деоксиаденозин три-фосфат ингибируют различные клеточные процессы, включая
активность рибонуклеоитидной редук-тазы, что приводит к смерти клетки и
повреждению тканей. Активность эритроцитарной ADA, как пра-вило, измеряется при
постановке диагноза ADA дефицита, но обычно оказывается ниже, чем в лим-фоцитах.
Клинический и лабораторный спектр дефицита ADA довольно широк и зависит от
тяжести лежащих в основе генных мутаций. Скрининг большой группы здоровых
взрослых показал, что 7% или более нормальной активности эритроцитарной ADA
ассоциирует с интактным иммунитетом; поэтому SCID и менее тяжелые иммунные
дефекты при дефиците ADA связаны с мутациями в гене ADA, которые устраняют или
почти полностью выключают функцию энзима. Приблизительно у 80% пациентов име-ет
место его раннее начало, классический дефицит ADA и развитие в первые 3 месяца
жизни. Отли-чительной клинической характеристикой у приблизительно 50% этих
пациентов являются скелетные аномалии, первичные образование чашеобразных
углублений (cupping) и постепенное сведение на нет или распространение наружу
(flaring) костохондральных соединений видимых на рентгенограмме грудной клетке.
Эти пациенты сохраняют 0,01% или менее активности ADA, у них имеется
алимфоци-тоз (Приблизительно от 15% до 20% случаев дефицита ADA не диагносцируются вплоть до
возраста 1-2 года; эти пациенты классифицируются, как имеющие отсроченное
позднее начало SCID вызванным менее разрушительными мутациями в гене ADA.
Пораженные дети сохраняют 0,1% до 2% активности ADA, количество периферических
лимфоцитов крови оказывается менее 500 клеток на мм3, но менее тяжелую
гипоальбуминемию на первом году жизни; тем не менее, снижение числа Т и В клеток
и их функций происходит довольно быстро и вскоре после этого развивается
инфекция. Позднее начало дефицита ADA имеет место у 5% и менее пациентов и
характеризуется диагнозом комбиниро-ванного иммунодефицита между 3 и 15 годами
жизни, которому обычно предшествует история перситирующей герпетической инфекции
и рекурентных инфекций верхних дыхательных путей, часто Streptococcus pneumoniae.
Аутоиммунные заболевания, особенно гемолитическая анемия и
тромбоцитопенияассоциируют с поздним началом ADA дефицита. Лабораторные данные
отличительны и включают 2% до 5% актив-ности ADA, количество циркулирующих
лимфоцитов менее, чем 800 клеток/мм3, количество CD3+ T-клеток менее, чем 500
клеток на мм3 и эозинофилию. Может развиться гипогаммаглобулинемия с
от-сутсвтием IgG2 и повышенным уровнем в сыворотке IgE. In vitro Т-клеточная
функция и специфиче-ские реакции антител, в частности, на полисахаридные
антигены, значительно снижены. Были описа-ны изолированные семьи с началом во
взрослом возрасте и частичным ADA дефицитом.
Измерения активности ADA эритроцитов не надежно у пациентов получавших
трансфузии; вместо этого должна измеряться функция энзима в лимфоцитах или
фибробластах для подтверждения диаг-ноза ADA дефицита. Пренатальный диагноз ADA
дефицита осуществляется путем скрининга на ак-тивность энзима в клетках
полученных от плода, но должен сопровождаться для подтверждения сек-венсией ДНК.
У иммунодефицитарных пациентов было идентифицировано более 60 различных мута-ций.
Большинство из них кластировано в секвенциях ДНК кодирующих амино кислоты
участвующие в связывании субстрата или обладающих каталитическими функциями.
Более, чем 50% мутаций у па-циентов с дефицитом ADA полностью выключают
активность энзима и проявляются ранним началом SCID. В отличие от других форм AR
SCID, при которых просматривается тенденция пациентов к го-мозиготности в
отношении одной и той же мутации, большинство ADA дефицитных пациентов являются
смешанно гетерозиготными в отношении двух различных мутантных ADA аллелей.
Оптимальной терапией ADA-дефицитного SCID является ткане-совместимая BMT.
Приживление гап-лоидентичной BMT, даже с проведением претрансплантарной аблации,
кажется сниженным при ADA дефиците по сравнению с другими диагнозами SCID.
Уровень смертности также может оказаться уве-личенным. По этим причинам
гаплоидентичная BMT обычно не проводится для лечения ADA дефици-та; вместо этого
пораженные пациенты при отсутствии потенциального ткане-совместимого донора
часто лечатся терапией замещения ADA. Замещение энзима не требует захвата ADA в
лимфоциты, чтобы оказать благоприятный эффект на Т и В клеточные функции.
Токсические метаболиты присут-ствуют в экстрацеллюлярной жидкости и пребывают в
равновесии с интрацеллюлярными продуктами; поэтому снижение уровня метаболитов в
плазме путем их деградации парентерально вводимой ADA приводит к снижению
концетрации внутриклеточных метаболитов. Полиетилен-глюкол связаннный
(PEG)-говяжья ADA применяется с 1986 года и дает частичную иммунную
реконституцию и длитель-ное улучшение состояния и выживаемость пораженных
пациентов. Пациенты с ранним началом SCID обычно получают от 30 до 60 U/kg/в
неделю PEG-ADA; доза может снижаться у детей с возрастом. Недостатками PEG-ADA
терапии является высокая стоимость и необходимость частых введений и тщательного
мониторинга уровня метаболитов.
Реконституция иммунитета в детей получающих PEG-ADA не полная, с улучшением
числа В клеток и функции, чем таковых Т клеток. Пациенты остаются лимфопеничными
(ADA дефицит был первым нарушением лечащимся генной терапией гематопоетических
клеток. Неко-торые ADA-дефицитарные пациенты, включая трех новорожденных,
получали ADA ген-корректированные аутологичные зрелые Т лимфоциты или
костномозговые или пупочного канатика стволовые клетки. Хотя и казалось, что ADA
корректированные лимфоциты имеют селективные преимущества роста над
ADA-дефицитарными клетками, ни один из пациентов не был излечен от SCID или
прекратил полностью получение PEG-ADA терапии. Запланированы новые исследования,
векторный дизайн и неадекватная трансдуктивная эффективность человеческих
гематопоетических стволовых клеток остаются существенными препятствиями в
достижении успеха генной терапии дефицита ADA.
АТИПИЧНЫЙ ТЯЖЕЛЫЙ КОМБИНИРОВАННЫЙ ИММУНОДЕЦИТ И КОМБИНИРОВАННЫЕ ИММУНОДЕФИЦИТЫ
Дефицит ZAP -70
Дефицит ZAP -70 - это редкий AR SCID , который был впервые описан в 1994 году среди отдельных семей близкородственных родителей. Лимфоцитоз (4000-20 000 клеток на мм 3), избыток (55-75%) CD 3+ CD 4+ и менее, чем 5% CD 8 + характерны для этой новой формы T + B + SCID . У большинства пораженных пациентов выявляется пониженный уровень IgG сыворотки, нарушенные В-клеточные реакции и отсутствие Т-клеточной функции, включая неспособность отторгать аллогенный кожный трансплантат ребенком.
Отсутствие CD 8 + T клеток связано с мутациями в протеине тирозин киназе, ZAP -70. Функция ZAP -70 является критической в проведении сигнала от TCR и его отсутствие проявляется ненормальной дифференцировкой тимоцитов и дефективной Т-клеточной активацией, необходимой для Т-клеточной пролиферации и функции. К настоящему времени описано менее 15 ZAP -70 дефицитных пациентов, у большинства из них имеются мутации в небольшой части ZAP -70 гена, которые поражают стабильность протеина и каталитическую функцию. Секвенсирование ДНК необходимо для подтверждения наследственности двух мутантных ZAP -70 аллелей у пациентов с SCID , характеризующихся бедностью циркулирующих CD 8 + T клеток.
Дефициты CD 3
TCR собираются и экспрессируются на поверхности в ассоциации с CD 3, который является комплексом из шести субединиц (eg, ed и xx). Две формы CD 3 дефицита были описаны в отдельных семьях, в которых они были вызваны мутациями в CD 3 протеине, что проявлялось дефективной экспрессией TCR . Хотя не так тяжелы как дефекты в рекомбинациях, мутации в CD 3g и CD 3e проявляются от слабого до умеренного иммунодефицитами. Пораженные пациенты имеют пониженное количество Т-клеток и их функции; В-клетки поражаются в различной степени.
Чистые лимфоцитарные синдромы
Чистые лимфоцитарные синдромы (BLS ) напоминают селективные Т-клеточные иммунодефициты, но у некоторых пациентов они не отличимы от таковых SCID . Два типа BLS отражают дефицитарную экспрессию основного комплекса ткане-совместимости (МНС) класс (HLA A , B C ) или класс II (HLA DR , DQ , или DP ) молекул на гематопоетических клетках. Диагноз типа 1BLS или MHC класс I дефицита предполагается, когда HLA A , B и C молекулы на лимфоцитах не могут быть определены серологическими методами. Это нарушение было идентифицировано среди небольшого числа семей с близкородственной связью и их клиническое проявление варьирует. В отличие от типа 2 BLS большинство пациентов с типом 1 BLS в детстве асимптоматичны; хотя персистирующие респираторные инфекции и хронические заболевания легких развиваются у этих детей в течение первых десяти лет жизни. Диагноз запаздывает, так как у пациентов сохраняется нормальное количество периферических T и В клеток и относительно сохраненные иммунные функции. Лимфоцитарный фенотип может выявить снижение CD 8+T клеток.
Тип 1 BLS связан с мутациями на одном из нескольких определенных генов. У некоторых пациентов имеются дефекты в транскрипции MHC класс 1 генов, в то время как у других отмечаются мутации в генах кодирующих транспортер ассоциированный с антигенным процессингом (ТАР)-1 или ТАР-2. Эти протеины участвуют в транспорте процессированных антигенов с цитоплазмы в эндоплазматический ретикулум. При отсутствии ТАР-1 или ТАР-2 МНС класс I молекулы в эндоплазматическом ретикулуме оказываются неспособными нагружаться антигенами, что проявляется в их деградации и снижении -клеточно-поверхностной экспрессии. Лечение типа 1 BLS является поддерживающим и ВМТ обычно не показана.
Тип 2 BLS или МНС класс II дефицита довольно редкая форма AR T + B + SCID , который в первую очередь поражает детей рожденных в близкородственных семьях североафриканского или средиземноморского происхождения. У пациентов с MHC класс II дефицитом часто развивается заболевание печени ассоциированное с хронической Cryptosporoium инфекцией. У пациентов отмечается нормальное количество циркулирующих лимфоцитов с уменьшением количества CD 4 + T клеток, гипогаммаглобулинемия и существенно дефективная in vitro Т-клеточная и В-клеточная пролиферация на специфические антигены.
Отличительной характеристикой типа 2 BLS является сниженная способность лимфоцитов стимулировать аллогенные лимфоциты в культурах in vitro . Это наблюдение подкрепляется обнаружением того факта, что МНС класс II молекулы экспрессируются с менее, чем 5% от нормальной интенсивности на гематопоетических клетках пораженных пациентов; тем не менее, анализ секвенсии ДНК не выявил мутаций в MHC класс II геназ; скорее у пациентов имеются наследственные мутации в одном из нескольких генов имеющих большое значение в их транскрипции, особенно CIITA , RFX -5, RFX -B или RFX -AP . Отдаленный прогноз для пациентов с типом 2 BLS катастрофичен. Большинство пациентов погибает от прогрессирующей органной недостаточности. Как правило, ткане-совместимая или гаплоидентичная ВМТ не дают иммунной реконституции и продление выживаемости при переходе во взрослый возраст.
Дефицит пурин нуклеозид фосфорилазы
Дефицит пурин нуклеозид фосфорилазы (PNP ) - редкий AR комбинированный иммунодефицит ассоциирующий с иммунными дефектами и нейрологическими симптомами, включая значительное отставание в развитии, проблемы с поведением и моторные отклонения. SCID с нейрологическими дефицитами должен рассматриваться как PNP дефицит до тех пор, пока не будет доказано иное. Клинические проявления PNP дефицита в основном такие же как при SCID ; однако, PNP часто классифицируется как комбинированный иммунодефицит, так как В-клеточные дефекты относительно слабо выражены в раннем детстве. Обычно у пациентов отмечается лимфопения (in vitro Т-клеточная функция. Снижение числа В-клеток и их функции происходит со временем, проявляясь в низких уровнях сывороточных IgG и IgA . Несмотря на существенное снижение В-клеточной функции, у более, чем 30% пациентов развиваются аутоиммунные заболевания, такие как гемолитическая анемия, тромбоцитарная пурпура и васкулит. Некоторые пациенты погибают от лимфомы и других опухолей.
PNP сопровождает ADA и предшествует гипоксантин фосфорибосилтрансферазе на пути расщепления пуринов. Она катализирует фосфорилирование инозин-деоксиинозина и гуанозин-деоксигуанозина до гипоксантина и гуанина, соответственно. Гуанозин и деоксигуанозин кажется являются токсичными для Т лимфоцитов, а пониженный уровень внутриклеточного гуанозин трифосфата (GTP ) может оказаться в первую очередь повреждающим ЦНС. Отсутствие функции PNP типично проявляется уровнем мочевой кислоты сыворотки менее, чем 1mg /dL , что может применяться для скрининга этого нарушения. Измерения эритроцитарной активности PNP служит для подтверждения диагноза. Секвенсирование ДНК выявило наследственность отдельных мутаций в большинстве семей. Многие пациенты с дефицитом PNP были рождены от родителей-родственников и у них имеются мутации полностью исключающие экспрессию протеина и энзиматической активности. Прогноз в отношении PNP дефицита чрезвычайно неблагоприятен. Только у небольшого количества пациентов успешно завершилась трансплантация гематопоетических стволовых клеток при BMT . GVHD является существенным осложнением и улучшения нейрологических симптомов после ВМТ не происходит. Энзим-заместительная и генная терапия при дефиците PNP в настоящее время не проводятся.
Атаксия телеангиектазия и варианты
Атаксия телеангиектазия (АТ) - это комплексное AR нарушение, характеризующееся церебральной атаксией, окулокутанной телеангиэктазией, клеточной радиочувствительность, предрасположенностью к злокачественным образованиям и комбинированным иммунодефицитом. Распространенность в мире АТ считается 3 на 10 6 живых родов. АТ мутированный генный (АТМ) продукт был идентифицирован в 1995 году и является сигнальной протеин киназой принимающей участие в контроле цикла-клетки, рекомбинации ДНК, апоптозе и других клеточных реакциях на повреждение ДНК.
Основным симптомом при АТ является нейрологическое нарушение, обычно проявляющееся в виде атаксической походки на втором году жизни, но наблюдаемой у более, чем 84% пациентов в возрасте 4 лет. Атаксия прогрессивно поражает туловище, конечности и палатальные мышцы, что проявляется в дизартрической речи, слюнотечением, окулярной апраксией, хореоатетозом и неспособностью к свободному передвижению к 10 году жизни. Телеангиектазия склер и кожи впоследствии развивается в возрасте между 4 и 8 годами. Клеточный и гуморальный иммунитет при АТ варьирует, но часто приводит к рекурентным инфекциям верхнего и нижнего респираторных трактов и хроническому заболеванию легких. Повышенная частота бактериальных и вирусных инфекций отмечается у большинства АТ пациентов между 3 и 6 годами жизни и несколько снижается при IVIG терапии. Важной чертой АТ является выраженная предрасположенность к Т-клеточным и В-клеточным новообразованиям, в частности лейкемии и лимфомам Hodgkin и non -Hodgkin . Злокачественные новообразования - это вторая наиболее частая причина смерти среди пациентов с АТ, впереди только легочные заболевания и аспирационная пневмония.
Генетическая нестабильность, проявляющаяся как хромосомальные транслокации и чрезмерные разрывы ДНК, является основным признаком АТ и может быть причиной рака и иммунодефицита у пораженных детей. Дополнительными клиническими проявлениями АТ являются задержка росте, гипогонадизм и позднее наступление пубертата и инсулин-резистентный некетонический сахарный диабет. Повышенный уровни в сыворотке a-фетопротеина (AFP ) и карциноэмбриотического антигена довольно часты и могут принести пользу при постановке диагноза.
У АТ пациентов имеется прогрессивная лимфопения, поражающая как Т, так и В клетки, включая селективную утрату CD 4 + T клеток с инверсией нормального соотношения CD 4-CD 8. Снижение как Т-клеточной, так и В-клеточной функции развивается со временем, и как считается, связано с чрезмерным образованием разрывов ДНК внутри гена TCR и иммуноглобулина на хромосоме 7 и 14, соответственно. АТ пациенты проявляют пониженную in vitro Т-клеточные пролиферативные реакции, но тяжесть иммунной дисфункции варьирует. Гуморальные дефекты включают IgA и IgE дефициты у большинства АТ пациентов и пониженные уровни изогемагглютининов и сывороточного IgG 2 у меньшего количества индивидуумов. Также могут быть нарушены специфические реакции антител.
Повышенная чувствительность к ионизирующей радиации и генетическая нестабильность довольно часты среди пациентов с АТ. АТМ может действовать активирующе на ориентиры клеточного цикла в ответ на повреждение ДНК, включая нормальные V )D )J рекомбинационные проявления в Т и В клетках. Отклонения в АТМ-опосредованном контроле клеточного цикла не мешают продолжению синтеза ДНК, несмотря на повреждения ДНК, что проявляется в накоплении хромосомальных обломков с течением времени и смертью клеток. Тимоциты, незрелые В лимфоциты и клетки ЦНС и васкулярного эндотелия могут быть более чувствительны к этим явлениям.
Большинство АТ пациентов являются гетерозиготным в отношении двух различных АТМ мутаций поражающих стабильность протеина. К сожалению, клеточная радиочувствительность не позволяет применять трансплантацию в качестве жизненной опции лечения АТ. Более того, стандартные протоколы химиотерапии не могут применяться для лечения ассоциирующих с АТ злокачественных новообразований. Некоторые АТ пациенты выживают в течение нескольких лет с лимфоидными опухолями, но отдаленные прогнозы катастрофичны. Большинство пациентов с АТ не переживают за третью декаду жизни. АТ гетерозиготные также могут быть предрасположены к опухолям.
К вариантам АТ относятся синдром разрывов Nijmegen (NBS ), который также характеризуется Т-клеточными и В-клеточными иммунными дефектами, радиочувствительностью, генетической нестабильностью и предрасположенностью к раку. NBS отличается от АТ наличием микроцефалии у более, чем 75% пораженных детей, умеренным отставанием умственного развития и фациальным дисморфизомом, отсутствием телеангиоектазии, прогрессивной атаксии и повышенных уровней AFP . Мутированный ген у пациентов с NBS кодирует Nibrin , протеин, подобный АТМ, участвующий в восстановлении ДНК и контроле клеточного цикла. Иммунодефицит у пациентов с NBS довольно значителен и включает лимфопению, уменьшение количества CD 4 + клеток и инверсию соотношения CD 4-CD 8, повышенное количество NK клеток и снижение in vitro Т-клеточной функции. Низкие или отсутствующие в сыворотке IgG и IgA при нормальном или повышенном уровне IgM встречается у более, чем 90% пациентов. IgG 2 дефицит и дефективная выработка пневмококковых антител также могут развиться.
Синдром Wiskott -Aldrich
Wiskott -Aldrich синдром (WAS ) - это Х-связанный наследственный иммунодефицит, характеризующийся экземой, врожденной тромбоцитопенией с мелкими тромбоцитами и рекурентными инфекциями. Он поражает приблизительно от 1 до 2 из 10 6 детей. Продукт WAS гена был идентифицирован как WAS протеин (WASP ) в 1994 году. Наследственная Х-связанная тромбоцитопения (XLT ) также, как было впоследствии показано, развивается в результате мутаций в WASP . WASP - это внутриклеточная сигнальная молекула, которая через связь с другими протеинами участвует в проведении TCR сигнала, играя важную роль в регуляции действия цитоскелетной организации.
Строгая корреляция существует между генотипом и фенотипом при WAS . Хотя у всех пациентов имеются отклонения в тромбоцитах, лица мужского пола с наиболее тяжелыми мутациями в гене WAS (составляющие приблизительно 30% всех случаев) находятся под наибольшим риском смерти от кровотечения, инфекции, аутоиммунитета или злокачественных образований. Пациенты с XLT или слабовыраженным WAS могут иметь экзему и различный иммунодефицит, но рак и аутоиммунные заболевания у них не развиваются. Пациенты с тяжелым или классическим WAS часто дают в периоде новорожденности или раннем детстве петехии и кровотечения вызванные тромбоцитопенией (обычно 10 000-50 000 клеток на мм 3). Кровавая диарея, внутричерепные кровоизлияния и чрезмерная кровоточивость из остатка пупочного канатика или после циркумцизии иногда оказываются изначальными проявлениями классического WAS . Диагноз может быть подтвержден определением размеров тромбоцитов. В отличие от идиопатической тромбоцитопенической пурпуры тромбоциты при WAS имеют средний объем от 3,8 до 5.0 fL (интервал нормы от 7,1 до 10,5 fL ). Хотя WAS не реагируют на стероиды или IVIG , тем не менее, спленектомия может проводится при тяжелой тромбоцитопении, но она повышает риск смерти связанной с сепсисом; тем не менее, впоследствии возникают кризы, подобные таковым при острой идиопатической тромбоцитопенической пурпуре, которые могут проявляться жизнеугрожающим кровотечением. Приблизительно 25% не связанных с BMT смертей при WAS вызвано осложнениями, связанными с кровотечением.
Экзема развивается у более, чем 80% пациентов и часто встречается до 6 месяцев жизни. Рекуррентные сино-пульмональные инфекции инкапсулированными микроорганизмами развиваются у мальчиков с классическим WAS в течение первых 2 лет жизни. Менее тяжелые инфекции можно наблюдать у пациентов с XLT или слабым WAS . Со снижением T -клеточной функции становятся проблематичными оппортунистические инфекции такие, как пневмония, вызванная Pneumocystis carinii и рекурентные инфекции вызванные вирусом герпеса. Приблизительно от 40% до 50% смертей, не связанных с BMT , при WAS вызваны инфекциями.
У пациентов с классическим WAS , не подвергавшимся трансплантации, существенно увеличена распространенность лейкемии, лимфом области живота и ЦНС и опухоли, ассоциирующие с вирусом Ebstein -Barr (EBV ), составляя 25% всех случаев смерти, не связанных с BMT . Аутоиммунные заболевания, включая гемолитическую анемию, артриты, васкулиты, воспалительное заболевание толстого кишечника и гломерулонефрит, наблюдаются у приблизительно 40% пациентов с тяжелыми WASP мутациями, не подвергавшимся трансплантации, и могут усиливать риск последующих злокачественных новообразований. Злокачественные новообразования и аутоиммунитет не встречаются у пациентов с XLT или слабым WAS .
У пораженных пациентов в детстве определяются нормальные количества лимфоцитов периферической крови. Лимфопения (CD3 + и CD 8 + Т клеток обычно наблюдается в 6-летнем возрасте. И, наоборот, количества В клеток и NK клеток остаются нормальными. У большинства пациентов развивается нарушение Т-клеточной функции. Нормальный уровень IgG сыворотки и пониженный IgM в ассоциации с дефективной выработкой пневмококковых антител и отсутствие изогемагглютининов постоянно встречается у WAS пациентов. Так как WAS пациенты дают нарушенную Т- и В-клеточные функции, то большинство из них получают IVIG .
Трансплантация гематопоетических стволовых клеток является единственным способом лечения классического WAS . Наибольший успех иммунной и тромбоцитарной реконституции достигается у детей получивших ткане-совместимую BMT до возраста 5 лет. Трансплантация стволовых клеток крови пупочного канатика и matched unrelated BMT также оказываются успешными у маленьких детей. Претрансплантаная аблация необходима для гарантии внедрения донорских Т клеток и тромбоцитов. Отторжение трансплантанта при гаплоидентичном BMT обескураживающе высоко, но пациенты с WAS прошедшие успешную трансплантацию излечиваются от иммунодефицита и других осложнений заболевания. Более, чем 100 мутаций при WASP было описано и многие из них уникальны для пораженных семей, тем не менее, некоторые горячие точки в гене WAS были идентифицированы, в которых с наибольшей частотой встречаются мутации. Классический WAS обычно вызывается мутациями, которые подавляют, исключают экспрессию протеина.
Х-связанное лимфопролиферативное заболевание
Х-связанное лимфопролиферативное (XLP ) заболевание или синдром Dunkan - это комбинированный иммунодефицит, который реализуется после экспозиции к вирусам Ebstein -Barr (EBV ). Частота XLP составляет приблизительно 1-3 на 10 6 мальчиков. XLP (LYP ) ген, как было показано в 1998 году, кодирует Т-клеточн-специфический протеин SAP (сигнальный лимфоцит активации молекула-ассоциированный протеин). SAP играет важную роль в регуляции проведения сигнала в активированных Т клетках, и SAP -дефицитные Т клетки не могут контролировать EBV -индуцированную В-клеточную пролиферацию; поэтому сниженная способность иммунной системы элиминировать EBV -инфицированные В лимфоциты при XLP связана с дефектами в EBV -специфичных Т-клеточных реакциях скорее, чем в В клетках.
До EBV -инфекции большинство мальчиков с мутациями в XLP клинически здоровы. Экспозиция к EBV проявляется фульминантным и часто фатальным инфекционном мононуклеозозом в 60% случаев. Тяжелый гепатит, вирус-ассоциированный гематофагоцитарный синдром, апластическая анемия и мультисистемная органная недостаточность сопровождаемые смертью развиваются в течение 8 недель после начала инфекционного мононуклеоза в 95% случаев. Hodgkin и non -Hodgkin лимфомы брюшной полости и ЦНС развиваются приблизительно у 30% мальчиков с XLP . Очень мало пациентов с EBV -ассоциированным лимфопролифератиным заболеванием выживает более 10 лет. Пациенты с дисгаммаглобулинемией, характеризующейся гипогаммаглоублинемией с повышеннм уровнем IgM в сыворотке, имеют благоприятный прогноз, если нет ассоциации с лимфопролифератинвым заболеванием. Прогноз для пациентов с XLP катастрофичен, при том, что большинство детей умрет в детском или подростковом возрасте. Антивирусные или иммуносупрессивные препараты оказываются не эффективными при лечении фульминантного заболевания. Только у небольшого количества пациента наступила иммунная реконституция после трансплантации; однако, трансплантация связана с возрастом пациента: большинство пациентов старше 15 лет не выживают после BMT .
После EBV инфекции у XLP пациентов выявляются Т-клеточные и В-клеточные дефекты. Хотя у пациентов и имеется нормальное количество Т-лимфоцитов, тем не менее, преобладают CD 8 + Т клетки с инверсией нормального соотношения CD 4-CD 8. In vitro Т-клеточная функция варьирует, но часто подавлена по сравнению с нормальными реакциями. Титры антител к EBV -ассоциированному нуклеарному антигену отсутствуют или существенно снижены во многих случаях, в то время, как выработка антител к капсид антигену варьирует. Отклонения функции NK проявлялась у 30% пораженных мальчиков.
СЕЛЕКТИВНЫЕ Т-КЛЕТОЧНЫЕ ДЕФЕКТЫ
Синдром DiGeorge
Больше чем 90% детей со сложными врожденными сердечными и краниофациальными пороками, которые вначале диагносцировались как отличающиеся различные генетические синдромы, включая синдромы DiGeorge (DGS ), Shprintzen ’s , велокардиальный и конотрункальная аномалия лица, как было показано, имеют общую микроделецию одной из копий хромосомы 22. Гемизиготная хромосомная 22q 11.2 микроделеция, как считается связана со многими медицинскими проблемами, с которыми сталкиваются пораженные дети, включая задержку развития и иммунодефицит. При анализе флюоресцентной in situ гибридизации (FISH ) эта хромосомная аномалия может быть диагносцирована у любого ребенка и у его или ее ранее не распознанных членов семьи.
DGS классически включает конотрункальные пороки сердца в ассоциации с персистирующей гипокальцемией и клеточным иммунодефицитом вторичным по отношению к более первазивному дефекту развития поля третьего и четвертого глоточного карманов, которые поражают паратиреоидные железы и тимус. Специфические аномалии сердца чаще всего ассоциирующие с DGS - это: прерывание аортальной дуги, тетральгия Fallot и truncus arteriosus . Фенотипическая экспрессия при DGS и других делеционных нарушениях хромосомы 22q 11.2 варьирует даже внутри семей и часто включает расщепление неба и фациальные аномалии.
Микроделеции хромосомы 22q 11.2 , выявляемые при проведении FISH , имеют различную величину, но обычно включают секвенцию относящуюся к так называемому критическому региону DiGeorge или локус DGS -1. В приблизительно 25% случаев делеция наследственна, часто получаемая от фенотипически здорового родителя. Остальные делеции хромосомы 22q 11.2 возникают de novo у пораженных детей. Распространенность этой хромосомной аномалии составляет, по крайней мере 13 случаев на 100 000 живых родов, что делает ее наиболее частой генетической причиной врожденного порока сердца; однако, 20% детей с синдромом делеции хромосомы 22q 11.2 не имеют сердечных аномалий. Анализ детального маппирования позволил определить минимальный DiGeorge критический регион, но специфический ген(ы) ответственный за хромосомы 22q 11.2 делеционный синдром не был обнаружен. Несмотря на это эти нарушения считаются как возникающие в результате гоплонедостаточности генов участвующих в нейральной перекрестной миграции. Вполне определенно можно сказать, что так как фенотипы проявляемые детьми с синдромом делеции хромосомы 22q 11.2 и их пораженными членами семьи включает широкий спектр физикальных находок, то множественные гены и модифицирующие факторы определяют их причину. Более того, редкие пациенты с фенотипическими проявлениями DGS не имеют определяемой микроделеции хромосомы 22q 11.2, вместо этого некоторые из этих пациентов являются гомозиготными по делеции хромосомы 10p , которая определяет DGS -II локусы или локус.
Спектр иммунных дефектов при DGS довольно широк, но большинство из них часто включает снижение количества (CD3+ T -лимфоцитов, количество СВ4+ Т-клеток менее 1000 клеток на мм 3 и умеренно нарушенный клеточный иммунитет. У большинства пациентов с DGS определяется 50% или менее от нормального количества Т-клеток и у 20% дают in vitro Т-клеточно пролиферативные реакции менее, чем 50% от нормы. Некоторые иммунологи полагают, что диагноз DGS должен ограничиваться детьми с синдромом хромосомальной 22q 11.2 делеции, у которых имеется менее, чем 500 клеток CD 3+ Т-лимфоцитов на мм 3 . Исследователи обычно считают, что созревание Т-клеток у пациентов с DGS в норме и, что лимфопения напрямую связана с количественным уменьшением функциональной массы тимуса. Никакие лабораторные исследования или фенотипические маркеры не коррелируют в достаточной степени с иммунологическим результатом. Обычно иммунный дефект улучшается с возрастом, и многие дети вырабатывают нормальные количества функциональных Т лимфоцитов к концу второго года жизни; однако, скорость, величина иммуннного восстановления не предсказуема и приблизительно у 5% пациентов с DGS имеются значительно сниженные количества Т-клеток и функции, как результат аплазии тимуса; этим пациентам может потребоваться BMT . У пятидесяти процентов пациентов отмечается также нарушение гуморального иммунитета. К последствиям иммунодефицита у детей с DGS относится повышенная предрасположенность к вирусным инфекциям. Это открытие проявляется на практике отсрочкой иммунизаций живыми вирусами до тех пор, пока не будут получены свидетельства иммунокомпетентности. Обычный критерий включает нормальное количество Т-клеток, 75% или более от нормы in vitro Т-клеточных пролиферативных реакций и специфическую выработку антител после иммунизации tetanus toxoid . Наличие и тяжесть иммунных дефектов у детей с синдромом делеции хромосомы 22q 11.2 варьируют и не коррелируют с другими фенотипическими проявлениями. Более того, Т-клеточный иммунодефицит развивается также часто у детей с диагнозом не-DGS хромосомальным 22q 11.2 делеционным синдромом, как и у детей с проявлением классического DGS .
До недавних пор многие дети с хромосомальным 22q 11.2 делеционным синдромом умирали вследствие их сердечных пороков. Эти дети сейчас выживают до взрослого возраста и появляются новые аспекты их хронических заболеваний. Признанными проявлениями хромосомального 22q 11.2 делеционного синдрома в настоящее время считаются аутоиммунитет, существенные проблемы с обучением, поведенческие и эмоциональные дисфункции и психиатрические диагнозы. Важно проводить консультацию семьи детально обрисовывая эти проявления хромосомального 22q 11.2 делеционного синдрома.
Хронический мукокутанный кандидиаз
Хронический мукокутанный кандидиаз (СМС) - это гетерозиготная группа нарушений характеризующаяся персистенцией фунгальной инфекции ногтей, кожи, и слизистых мембран. Более, чем 50% пациентов при этом имеют AR заболевание характеризующееся полиэндокринопатией и имеют мутации в гене AIRE кодирующем транскрпционный регуляторный протеин. В основе лежащий генетический дефект у детей с изолированным СМС не известен. У большинства пациентов имеется нормальное количество лимфоцитов и функция; однако, отличительным проявлением у некоторых пациентов с изолированным СМС является отсутствие in vitro Т-клеточной пролиферации на кандидальный антиген.
РЕЗЮМЕ
Т-клеточные иммунные дефекты включают большинство наследственных иммунодефицитов диагносцируемых в детском возрасте. Большинство клеточных иммунодефицитов ассоциирует с гуморальными дефектами при различных клинических и лабораторных проявлениях. В основе лежащие генетические дефекты известны и определены для большинства наследственных Т-клеточных иммунных дефектов и анализ мутаций все быстрее становится интегральной частью оценки и диагностики. Детальное обсуждение с семьей корреляции фенотипа-генотипа является критическим для медицинского ведения и отдаленного прогноза.
Источник: Melisa E. Elder, / T-CELL
IMMUNODEFICIENCIES / PEDIATRIC CLINICS OF NORTH AMERICA VOL.47. N 6.
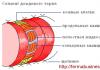
Т-клеточные рецепторы (англ. TCR) - поверхностные белковые комплексы Т-лимфоцитов, ответственные за распознавание процессированных антигенов, связанных с молекулами главного комплекса гистосовместимости (англ. MHC) на поверхности антиген-представляющих клеток. TCR состоит из двух субъединиц, заякоренных в клеточной мембране и ассоциирован с многосубъединичным комплексом CD3. Взаимодействие TCR с MHC и связанным с ним антигеном ведет к активации Т-лимфоцитов и является ключевой точкой в запуске иммунного ответа.
TCR представляет собой гетеродимерный белок, состоящий из двух субъединиц - α и β либо γ и δ, представленных на поверхности клетки. Субъединицы закреплены в мембране и связаны друг с другом дисульфидной связью.
По своей структуре субъединицы TCR относятся к суперсемейству иммуноглобулинов. Каждая из субъединиц образована двумя доменами с характерной иммуноглобулиновой укладкой, трансмембранным сегментом и коротким цитоплазматическим участком.
N-концевые домены являются вариабельными (V) и отвечают за связывание антигена, презентируемого молекулами главного комплекса гистосовместимости. В составе вариабельного домена содержится характерный для иммуноглобулинов гипервариабельный участок (CDR). За счет необычайного разнообразия данных участков, различные Т-клетки способны распознавать широчайший спектр различных антигенов.
Второй домен - константный (C) и его структура одинакова у всех субъединиц данного типа у конкретной особи (за исключением соматических мутаций на уровне генов любых других белков). На участке между С-доменом и трансмембранным сегментом имеется остаток цистеина, с помощью которого между двумя цепями TCR образуется дисульфидная связь.
Субъединицы TCR агрегированы с мембранным полипептидным комплексом CD3. CD3 образован четырьмя типами полипептидов - γ, δ, ε и ζ. Субъединицы γ, δ и ε кодируются тесно сцепленными генами и имеют близкую структуру. Каждая из них образована одним константным иммуноглобулиновым доменом, трансмембранным сегментом и длинной (до 40 аминокислотных остатков) цитоплазматической частью. Цепь ζ имеет маленький внеклеточный домен, трансмембранный сегмент, и большой цитоплазматический домен. Иногда вместо цепи ζ в состав комплекса входит цепь η - более длинный продукт того же гена, полученный путем альтернативного сплайсинга.
Поскольку структура белков комплекса CD3 инвариантна (не имеет вариабельных участков), они не способны определять специфичность рецептора к антигену. Распознавание является исключительно функцией TCR, а CD3 обеспечивает передачу сигнала в клетку.
Трансмембранный сегмент каждой из субъединиц CD3 содержит отрицательно заряженный аминокислотный остаток, а TCR – положительно заряженный. За счет электростатических взаимодействий они объединяются в общий функциональный комплекс Т-клеточного рецептора. На основании стехиометрических исследований и измерения молекулярной массы комплекса наиболее вероятным его составом является (αβ)2+γ+δ+ε2+ζ2.
TCR, состоящие из αβ-цепей и γδ-цепей весьма близки по структуре. Эти формы рецепторов по-разному представлены в различных тканях организма.
Структура рецептора Т-лимфоцита во многом напоминает структуру молекулы антитела. Молекулы Т-клеточных рецепторов (ТКР) состоят из двух цепей - а и р. Каждая из них содержит V- и С-домены, их структура закреплена дисульфидными связями. Вариабельные домены а- и р-цепей имеют не 3-4, как у антител, а не менее 7 гипервариабельных участков, которые формируют активный центр рецептора. За С-доменами, около мембраны, располагается шарнирная область из 20аминокислотных остатков. Она обеспечивает соединение а- и р-цепей с помощью дисульфидных связей. За шарнирной областью располагается трансмембранный гидрофобный домен из 22 аминокислотных остатков, он связан с коротким внутрицитоплазматичеким доменом из 5-16 аминокислотных остатков. Распознавание Т-клеточным рецептором представляемого антигена происходит следующим образом. Молекулы МНС классаП, как и рецепторы Т-лимфоцитов, состоят из двух полипептидных цепей - а и р. Их активный центр для связывания представляемых антигенных пептидов имеет форму «щели». Она формируется спиральными участками а- и р-цепей, соединенными на дне «щели» между собой неспиральной областью, образованной сегментами той и другой цепи. В этом центре (щели) молекула МНС присоединяет процессированный антиген и таким образом представляет его Т-клеткам (рис. 63). Активный центр Т-клеточного рецептора образуется гипервариабельными участками а- и р-цепей. Он также представляет собой своеобразную «щель», структура которой соответствует пространственной структуре представляемой молекулой МНС классаП пептидного фрагмента антигена в такой же степени, как структура активного центра молекулы антитела соответствует пространственной структуре детерминанта антигена. Каждый Т-лимфоцит несет рецепторы только для одного какого-то пептида, то есть специфичен в отношении конкретного антигена и связывает процессированный пептид только одного типа. Присоединение представляемого антигена к Т-клеточному рецептору индуцирует передачу сигнала от него на геном клетки.
Для функционирования любого ТКР необходим его контакт с молекулой CD3. Она состоит из 5субъединиц, каждая из которых кодируется своим геном. Молекулы CD3 имеют все субклассы Т-лимфоцитов. Благодаря взаимодействию Т-клеточного рецептора с молекулой CD3 обеспечиваются следующие процессы: а)вынос ТКР на поверхность мембраны Т-лимфоцита; б)придание соответствующей пространственной структуры молекуле Т-клеточного рецептора; в)прием и передача сигнала Т-клеточным рецептором после его контакта с антигеном в цитоплазму, а затем в геном Т-лимфоцита через фосфатидилинозитольный каскад с участием посредников.
В результате взаимодействия молекулы МНС классаП, несущей антигенный пептид, с рецептором Т-лимфоцита пептид как бы встраивается в «щель» рецептора, которую образуют гипервариабельные участки а- и р-цепей, контактируя при этом с той и другой цепью
Роль вилочковой железы - основного органа в иммунной системе определилась лишь в 1961 г. благодаря данным R. A. Good и J. F. Miller. Несмотря на наличие многочисленных сведений о роли вилочковой железы в созревании лимфоидной системы, многие стороны этой проблемы, в частности превращение незрелых претимических лимфоидных клеток в иммунокомпетентные Т-клетки со значительной функциональной гетерогенностью, окончательно не выяснены.
Т-клетки, прошедшие через вилочковую железу, приобретают способность реагировать на антигены и митогены (ФГА и КонА), расселяются в тимусзависимых зонах лимфоидных органов (в паракортикальных зонах лимфоузлов, периартериальных фолликулах селезенки) и составляют основную массу рециркулирующего пула в периферической крови с длительным жизненным циклом. Эти клетки получили название тимусзависимых.
Развитие Т-клеток
Внутритимусное развитие обеспечивается различными факторами: специфическим «микроокружением», определяемым стромой вилочковой железы, а также ее гормонами, эпителиальными и мезенхимальными клетками. Из вилочковой железы выделяют ряд активных веществ, отличающихся активностью молекулярной массой, устойчивостью к температурным воздействиям. Среди них описывают тимозин, тимопоэтин, тимический гуморальный фактор. Эти вещества, в частности тимозин, способствуют превращению Т-клеток в более зрелые формы.
Механизм действия тимозина и других активных веществ на Т-клетки до конца не ясен, полагают, что точкой приложения их является мембрана клеток, а именно аденилатциклазная система. Вероятно, тимозин активизирует ее и повышает концентрацию внутриклеточных цАМФ и цГМФ. Указанные нуклеотиды имеют отношение к процессам клеточного развития. Последние очень сложны, многообразны и требуют еще дальнейшего изучения.
Виды и функции Т-клеток
Среди Т-клеток выделяют несколько субпопуляций, отличающихся кортизолчувствительностью и различием антигенов и рецепторов на их поверхности.
По назначению Т-клетки являются ответственными главным образом за клеточный иммунитет, но по своим функциям они неоднородны. В их составе различают несколько подклассов: клетки, распознающие антиген; клетки-хелперы; клетки-киллеры; клетки-супрессоры; клетки памяти и др. Очень сложным и дискутабельным является вопрос о том, когда происходит деление Т-клеток на различные по функциональному назначению субпопуляции. Неясно, что определяет функциональную направленность развития, происходят ли эти субпопуляции из единого предшественника или различие генетически детерминировано еще на уровне стволовой клетки.
Все субпопуляции Т-клеток составляют около 60-70% всех лимфоцитов; около 25% - В-клетки, или бурсозависимые, прошедшие «обучение» в другом центральном органе иммунитета - фабрициевой сумке (у птиц) или ее аналогах у человека, где приобретают отличительные от Т-клеток свойства. Следует отметить, что длительные поиски этого аналога не привели к успеху. Эту роль отводят различным лимфоидным образованиям - червеобразному отростку, миндалинам, групповым лимфатическим фолликулам кишок, лимфоидным скоплениям в легких и других органах. После созревания В-лимфоциты перемещаются преимущественно в фолликулы селезенки и лимфоузлы.
Большинство антигенов являются тимусзависимыми, поскольку иммунній ответ на них идет с обязательным участием Т-клеток; эти антигены комплексные, имеют несколько антигенных детерминант с различной специфичностью (тканевые, микробные, вирусные).
Механизм антигенного распознавания Т-клетками очень сложен, окончательно не изучен. Основной задачей Т-клеток и их рецепторов является распознавание «своих» и «чужих». антигенов. Этиология клеточных рецепторов Т-клеток, распознающих антигены, окончательно не установлена. Взаимодействие клеточных рецепторов с антигеном является сигналом для начала иммунного ответа и клеточной дифференцировки. Если иммунный ответ идет по типу антитело- образования, то для участия В-клеток и их трансформации в плазмоциты необходимо участие Т-клеток-хелперов (Th). Развитие и клеточных реакций, осуществляемых Т-клетками, происходит также с участием помощников - Т-ампфликаторов (Та).
Очень сложным и до конца не изученным является механизм хелперного действия Т-лимфоцитов (прямой контакт, или выделяемые клетками активные вещества). Активированные Th вырабатывают специфические факторы и через макрофаги активируют В-клетки. Сущность этих факторов до конца не определена. Учитывая, что В-клетки продуцируют различные классы иммуноглобулинов, не исключено существование разных субпопуляций Th для регуляции антителообразования (IgG, IgE).
Gershon в 1969 г. разработал концепцию о супрессорной роли Т-клеток (Т-супрессоры), затем было установлено, что Ts способны регулировать различные фазы иммунного ответа. Роль супрессоров заключается в регуляции уровня и силы специфического иммунного ответа, обеспечении иммунологической толерантности к некоторым тимусзависимым антигенам и к антигенам своих тканей. Ts несут рецепторы к IgG, IgM, действуют через растворимые медиаторы. Природа этих факторов, секретируемых Ts, активно изучается. Нет единого мнения по вопросу о механизмах, индукции Ts (прямой контакт клеток с антигеном или сигналы «обратной связи»).
Значение Ts очень велико, ограничение иммунной реакции не менее важно для организма, чем ее стимуляция. Это особенно касается тех иммунных реакций, которые могут привести к патологии (аутоиммунные болезни, заболевания)
Т-клетки-киллеры осуществляют реакции клеточного иммунитета: цитопатогенное действие при трансплантационном иммунитете, противоопухолевом, некоторых видах инфекций. Субпопуляция киллеров является и эффектором гиперчувствительности замедленного типа (ГЗТ).
Гетерогенностью Т-клеточной популяции по их функциональным свойствам объясняется чрезвычайно сложное взаимодействие клеток в реакциях организма на антиген.
В настоящее время очевидно, что не только Т-клетки, но и В-клетки также не однородная масса клеток. Для В-клеток характерно наличие на поверхности иммуноглобулинов, обеспечивающих распознавание антигенов, под влиянием которых В-лимфоциты превращаются в плазмоциты, продуцирующие антитела - . Выработка их на большинство антигенов не может осуществляться без взаимодействия с Т-клетками. Клеточная кооперация является необходимым условием для развития иммунного ответа.
Активированные Т-клетки кроме факторов, выделяемых Ts и Th, образуют ряд медиаторов - лимфокинов, которые имеют различные физико-химические свойства. Их роль — вовлечение в иммунный ответ различных клеток - нейтрофильных гранулоцитов, макрофагов, эозинофильных гранулоцитов. Участие этих Т-клеток в иммунном ответе очень важно. Особенно велика роль макрофагов, которые разрушают антиген и переводят его в иммуногенную форму. К макрофагам в настоящее время относят группу клеток, способных к фагоцитозу, прилипающих по стеклу - adherens, фагоцитирующие мононуклеары, тканевые макрофаги и моноциты.
Статью подготовил и отредактировал: врач-хирургТ-клеточный лейкоз-лимфома взрослых
Т-клеточный лейкоз-лимфома взрослых – это опухоль из CD4-лимфоцитов, вызванная относящимся к семейству ретровирусов Т-лимфотропным вирусом человека типа 1 (HTLV – Human T-lymphotropic virus) и характеризующаяся поражением кожных покровов и внутренних органов, резорбцией костной ткани, гиперкальциемией и наличием атипичных лимфоцитов в крови.
Чаще всего случаи данного заболевания регистрируются на юге Японии, реже на побережье Тихого океана, островах Карибского бассейна, в странах Экваториальной Африки, Южной Америки и на севере США. В основном болеют лица монголоидной и негроидной расы, причём более подвержены заболеванию представители мужского пола.
При Т-клеточном лейкозе-лимфоме опухолевые клетки по своей природе представляют собой активированные вирусом CD4-лимфоциты. Согласно статистике, развивается такое состояние в среднем у 5% инфицированных, у остальных лиц имеет место носительство провируса в CD4-лимфоцитах. В этой связи среди учёных актуально мнение о том, что в патогенезе данного заболевания участвуют ещё и другие факторы (возможно, генетическая предрасположенность и влияние окружающей среды), на фоне которых после заражения какая-то часть CD4-лимфоцитов обретает способность к неконтролируемому размножению. Отмечается повышенная митотическая активность, а вместе с ней дефицит клеточного иммунитета и накопление генетических дефектов.
Клинически опухоль даёт о себе знать генерализованным увеличением лимфатических узлов, гепатоспленомегалией (увеличением размеров печени и селезёнки), остеолизом (разрушением костей) и поражением кожи в виде опухолевидных образований, папул, бляшек, изъязвлений. Характерна также гиперкальциемия и повышение активности лактатдегодрогеназы в сыворотке крови. Инфильтрация костного мозга, как правило, совсем незначительна, тромбоцитопения и анемия не характерны.
Острая форма Т-клеточного лейкоза-лимфомы взрослых отличается неуклонным прогрессированием процесса и довольно низкой эффективностью лечения. Полихимиотерапия позволяет у значительной части (у 50-70%) больных добиться полной ремиссии, однако приблизительно у половины из них этот период не длится более одного года.
Глубокий иммунодефицит обусловливает очень высокую частоту вторичных инфекций. Многие из них провоцируются условно-патогенными микроорганизмами, вызывающими заболевания при резком снижении иммунитета.
На сегодняшний день описана также и хроническая форма заболевания, протекающая с поражением кожи, но без увеличения лимфатических узлов и гепатоспленомегалии. В этом случае характерен умеренный лимфоцитоз с невысоким содержанием опухолевых клеток в периферической крови. Продолжительность жизни таких пациентов может достигать и нескольких лет – до того момента, пока болезнь не трансформируется в острую форму.
Помимо острой и хронической, существует ещё две формы Т-клеточного лейкоза-лимфомы взрослых – это: лимфоматозная и тлеющая. Лимфоматозная развивается где-то у 20% больных. По клинической картине и своему течению данная форма во многом напоминает острую, отличаясь лишь малым числом атипичных лимфоцитов в периферической крови и выраженным увеличением лимфатических узлов.
Тлеющая форма встречается редко (не более 5%). Доля атипичных лимфоцитов в анализе крови здесь менее 5%. Гиперкальциемия, лимфаденопатия, гепатоспленомегалия, изменения со стороны костной ткани, центральной нервной системы и желудочно-кишечного тракта отсутствуют, лёгкие и кожа иногда поражаются. Продолжительность жизни обычно лет пять и более.
Необходимо отметить, что при любом варианте заболевания опухоль развивается именно за счёт моноклональной пролиферации CD4-лимфоцитов. Во всех этих клетках провирус встраивается в ДНК одинаково, обусловливая уникальную перестройку генов, которые кодируют антигенраспознающие рецепторы Т-лимфоцитов.
Т-клеточный крупногранулярный лимфоцитарный лейкоз встречается в 30-50 раз реже В-клеточного ХЛЛ и развивается в возрасте 50-55 лет, чаще у женщин. Основной морфологический признак заболевания - наличие больших гранулярных (содержащих азурофильные гранулы) лимфоцитов в периферической крови и костном мозге. Критерий диагностики - обнаружение больше 2 109/л больших гранулярных лимфоцитов в периферической крови. Наиболее частый иммунофенотип: CD3+, CD8+, CD4-, TCRab+.
Лейкемические клетки экспрессируют маркеры апоптоза (Fas или CD95 и Fas-лиганд), но резистентны к Fas-индуцируемому апоптозу.
Спленомегалия
выявляется у 20% пациентов, лимфаденопатия и гепа-томегалия встречаются еще реже. В связи с выраженной нейтропенией нередко возникают рецидивирующие инфекции. У 30% больных наблюдается ассоциация с аутоиммунными заболеваниями (аутоиммунная гемолитическая анемия, ревматоидный артрит и др.).
Течение заболевания вариабельно, стандартное лечение не разработано.
Т-клеточная лейкемия/лимфома взрослых
Т-клеточная лейкемия/лимфома взрослых - редкое лимфопролиферативное заболевание, встречающееся преимущественно в странах бассейна Карибского моря и Японии. Доказанным этиологическим фактором является ретровирус HTLV-1. В эндемичных районах инфицировано 5% населения; в течение жизни заболевает один человек из 50-100 инфицированных (в Японии при наличии около 1 миллиона вирусоносителей в год регистрируется около 500 случаев заболевания). Спорадические случаи Т-клеточной лейкемии/лимфомы взрослых отмечены в Европе и Северной Америке.
Опухолевые клетки полиморфны, с полисегментированными, похожими на лепестки цветка, ядрами. Иммунофенотип клеток: CD7-, CD2+, CD3+, CD4+, CD5+, CD25+. Наиболее частые цитогенетические находки - трисомия 12, del 6q.
В подавляющем большинстве случаев Т-клеточная лейкемия/лимфома взрослых характеризуется агрессивным течением, сопровождающимся анемией, лимфаденопатией, гиперкальциемией, ранней диссеминацией (поражение костей, кожи, центральной нервной системы), выраженным иммунодефицитом (обычно дисфункцией CD4+) с развитием тяжелых оппортунистических инфекций. В этих случаях прогноз неблагоприятный (медиана выживаемости не превышает 6 месяцев). Значительно реже встречаются более благоприятные варианты («тлеющий» и хронический), которые, однако, могут трансформироваться в агрессивный тип.
Грибовидный микоз